- Vim 编辑器:
- 生物信息学常见数据格式
- 文本处理三驾马车
- grep:
- grep结合正则表达式
- sed:
- -n :取消默认输出,只显示经过sed处理或匹配的行(常用)
- -e :直接在命令模式上进行 sed 的动作编辑,接要执行的一个或
- -i :直接修改读取的文件内容,不输出。
- 2 ∶ 第 2 行
- 2,4 ∶ 第 2 行到第 4 行
- 2,$ ∶ 第 2 行到最后一行,$ 表示最后
- 2~3 ∶ 从第 2 行开始,每隔 3 行取一行
- 2,+4 ∶ 从第 2 行到 2+4 行
- /pattern/ :匹配上 pattern 的行
- [!] : 表示否定,取反
- a ∶ append,在指定行的后增加一行,内容为 a 的后面接的字串
- i ∶ insert,在指定行的前增加一行,内容为 i 的后面接的字串
- d ∶ delete,删除某一行或者某几行,也可以指定删除匹配上的行
- c ∶ change,改变指定行的内容
- s ∶ 替换,使用格式为
- ‘s/pattern/new/[flags]’
- 把pattern替换
- /pattern/ :匹配上 pattern 的行
- cat readme.txt | sed ‘/www/ s/ee/EE/‘
- y ∶ 转换,实现字符一对一转换,格式 ‘y/inchars/outchars/‘
- cat readme.txt | sed ‘y/abcde/ABCDE/‘
- p ∶ print,把匹配或修改过的行打印出来,通常与–n参数合用
- cat readme.txt | sed -n ‘/ee/p’
- awk:
- 匹配结构:’ /pattern/{script}’
- awk内置变量:
- awk条件和循环语句:
- awk数学运算:
Vim 编辑器:
大多数Linux都会自带的文本编辑器。
功能强大:代码补全、编译及错误跳转等方便编程的功能特别
丰富,在程序员中被广泛使用。
功能强大到其官方现在对自己的定位是“程序开发工具”。
Vim 编辑器:
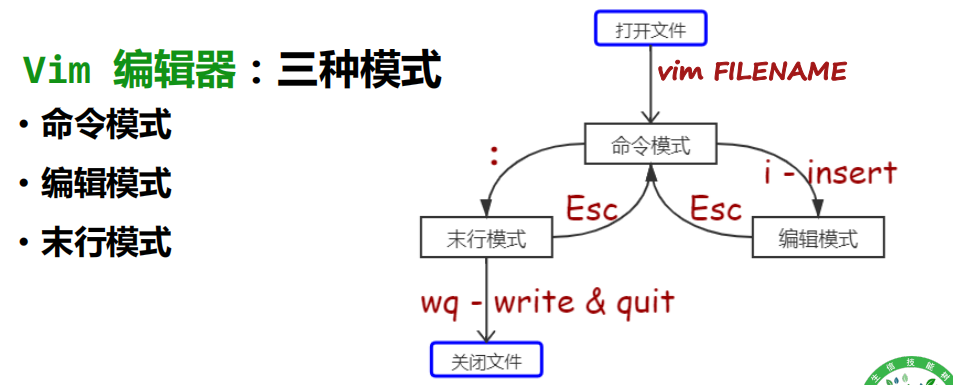
• 命令模式:
• 用vim FILENAME进入之后的默认模式
• 可以“上下左右”移动光标
• 可以剪切字符或整行、也可复制、粘贴
移动光标:
• 方向键或者hjkl
• ^和$:快速到所在行的开头和末尾(用0也可以到开头)
• 30j:向下移动30行(数字+方向进行快速移动)
• ctrl+f或b:上下翻页(forward/back)
• gg:快速回到文档的第一行的第一个字符
• G:快速到文件底部
剪切、复制与粘贴:
• x:剪切一个字符
• 10x:连续剪切10个字符(numberx)
• dd:剪切所在行
• 10dd:剪切所在行及向下共10行
• yy:复制光标所在行
• 10yy:复制所在行及下面10行
• p或P:在当前行的下面/上面进行粘贴
撤销动作(后悔药):
• u:复原前一个动作(可连续撤销)
• ctrl+r:重做上一个动作
• 编辑模式:
• 输入 i 后进入编辑模式(左下角有— INSERT —提示)
• 其实有多种进入的模式(i/a/o/I/A/O),但是记住一种即可
• 按esc键退出该模式
• 在编辑模式下即可正常的编辑、修改文本内容
• 末行模式:
• 在命令模式下输入一个 : 进入这一模式
• 注意需要是英文的冒号,中文的冒号(全角)输入不进来
• 按esc键退出该模式
• 在该模式下可以设置、查询、替换、保存并退出
保存与退出
• :wq 保存并退出
• :q 直接退出
• :q! 不保存修改,强制退出
• :wq! 保存并强制退出
查询
• 输入/KEYWORD进行查询
• 按n向下查找
• 按N向上查找
替换
• :%s///g 全局替换
• :s///g 替换光标所在行
• :s/// 只替换光标所在行的第一次
set系列命令
• :set nu 显示行号
• :set nonu 取消行号
• :set list 显示不可见字符(分辨制表符与空格)
• :set ff=unix 将文件格式转换为unix格式
帮助文档:
Vim 编辑器:vimtutor zh_CN 可以查看帮助文档
生物信息学常见数据格式
fasta
是一种基于文本用于表示核酸序列或多肽序列的格式。
其中核酸或氨基酸均以单
个字母来表示,且允许在序列前添加序列名及注释。
fastq
是一种存储了生物序列以及相应的质量评价的文本格式。
测序的原始序列。
gff
(General Feature Format)
记录序列中转录起始位点、基因、外显子、内含子等组成元件在染色体中的位置信息。
现在用得比较多的是第3版,即gff3。
gtf
(Gene transfer format)
可以用cufflinks里 的gffread命令互相
转换格式
文本处理三驾马车
grep:
一种强大的文本搜索工具,它能使用正则表达式匹配模式搜
索文本,并把匹配的行打印出来。
格式:grep [options] pattern file
常见参数:
-w:word 精确查找某个关键词 pattern
-c:统计匹配成功的行的数量
-v:反向选择,即输出没有没有匹配的行
-n:显示匹配成功的行所在的行号
-r:从目录中查找pattern
-e:指定多个匹配模式
-f:从指定文件中读取要匹配的 pattern
-i:忽略大小写
实际应用:
最常用的情况,用grep查参数
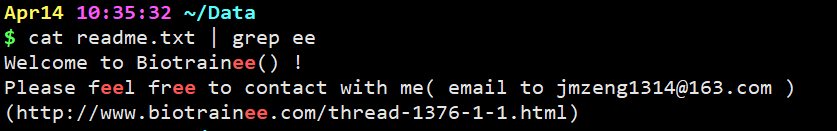
cat readme.txt | grep ee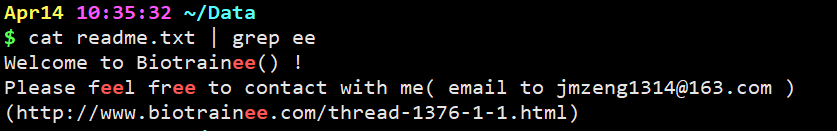
less example.gtf | grep -w -v exon
显示文件中不含exon的行
less example.fq | grep -c ‘@’
显示文件中含有@的行的数目
不能使用less example.fq | grep -w -c ‘@’,因为-w查找的是一个单词,@是一个字符,直接用-c就可以。
less example.gtf | grep -w -e CDS -e UTR
显示文件中含有CDS或者UTR的行
less example.gtf | grep -w -e CDS -e UTR | less -SN(加less -SN样式比较易读)
grep结合正则表达式
4.22日 linux生物信息学常见格式与grep 课程第 58分钟左右
提示:Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz文件就是
人类的Y染色体的gff文件
1.人类Y染色体上有多少个基因呢?
下面代码得出的结果把文件里的一些其他带有gene字样的也算进去了,不能这样。
zcat Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | grep -w ‘gene’ | less -SN
所以:
zcat Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | cut -f 3 | grep -w ‘gene’ | less -SN
意思是把文件的第三列提出来,在这一列中挑选有gene的。
zcat Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | cut -f 3 | grep -c ‘^gene’
意思是挑选出第三列以gene开头的,加“^”意思是以什么开头
2.在Y染色体的注释文件中有第三列哪些类型呢?
zcat Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | cut -f 3 | sort | uniq -c
先把文件的第三列提出来,然后排序,去重,显示数目。但是也有很多无用信息,怎么把无用信息去除呢?
zcat Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | cut -f 3 | grep -v ‘#’ | sort | uniq -c
这里添加了grep,可以把带#的行去掉
3、怎么把挑选的行提取出来?
zcat Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | grep -v ‘#’ | grep -w ‘exon’ > tmp
这里时把含有exon的行提取出来并且输出到tmp文件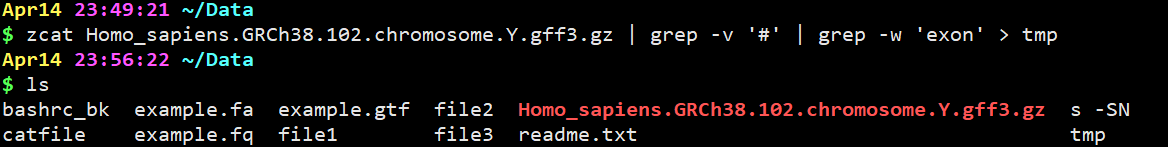
sed:
流编辑器,一般用来对文本进行增删改查
用法:
sed [-options] ‘script’ file(s)
常见参数:
-n :取消默认输出,只显示经过sed处理或匹配的行(常用)
-e :直接在命令模式上进行 sed 的动作编辑,接要执行的一个或
-i :直接修改读取的文件内容,不输出。
2 ∶ 第 2 行
2,4 ∶ 第 2 行到第 4 行
2,$ ∶ 第 2 行到最后一行,$ 表示最后
2~3 ∶ 从第 2 行开始,每隔 3 行取一行
2,+4 ∶ 从第 2 行到 2+4 行
/pattern/ :匹配上 pattern 的行
[!] : 表示否定,取反
常见 ‘script’ command :
a ∶ append,在指定行的后增加一行,内容为 a 的后面接的字串
cat readme.txt | sed ‘1a ahahhahahah’
意思是在readme.txt文件内容中的第一行后一行加上ahahhahahah字符串。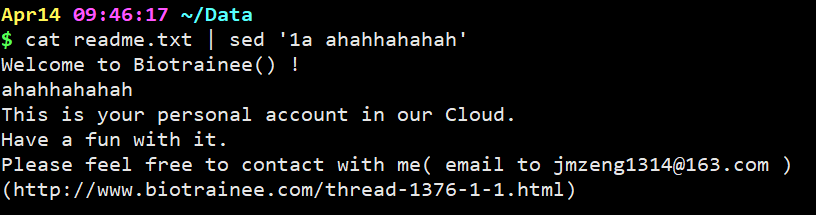
i ∶ insert,在指定行的前增加一行,内容为 i 的后面接的字串
cat readme.txt | sed ‘1,2a ahahhaahha’
意思是在第一行和第二行的后一行各加上字符串ahahhaahha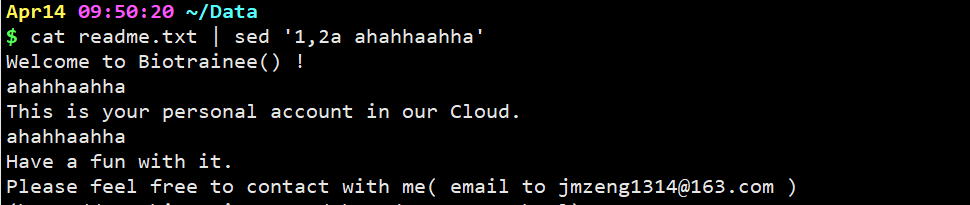
cat readme.txt | sed -e ‘1a lalallal’ -e ‘1i laalalla’
意思是在第一行的前面和后面分别加上一句lalallalla,用e进行多命令执行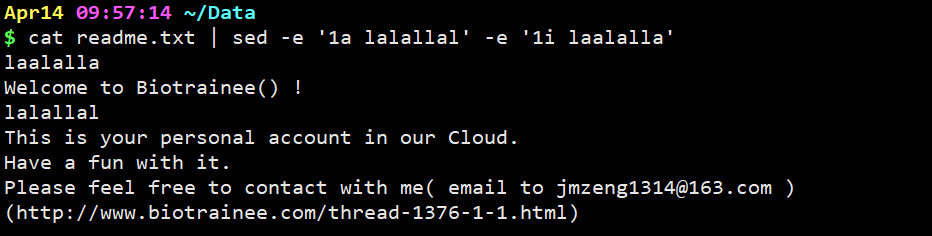
d ∶ delete,删除某一行或者某几行,也可以指定删除匹配上的行
c ∶ change,改变指定行的内容
cat readme.txt | sed -e ‘2,4i ########’ -e ‘2,4d’
意思是先在第二,三,四行的前面插入字符串######,然后再把原本的2,3,4行删掉。
cat readme.txt | sed -e ‘2c #######’ -e’3c #####’ -e ‘4c ########’
(笨方法,挨个改变)
cat readme.txt | sed ‘1,3!d’
意思是删掉除了1,2,3行之外的所有行,!代表取反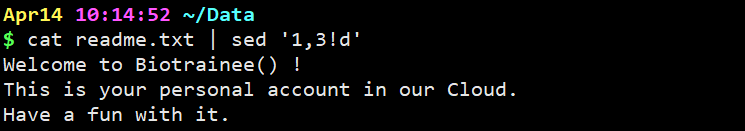
s ∶ 替换,使用格式为
‘s/pattern/new/[flags]’
把pattern替换
成new,默认只替换一个,可以指定flags
cat readme.txt | sed ‘s/o/%/g’
意思是把所有的o替换成%,g代表替换所有,不写的话默认只替换第一个,可以指定
cat readme.txt | sed ‘1,$ s/ee/EE/g’
把第一行到最后一行的所有ee替换成EE。
最后加上g,是替换所有的ee。如果不加g的话,例如在第四行中有两个ee,只能替换第一个。加了g就可以全部替换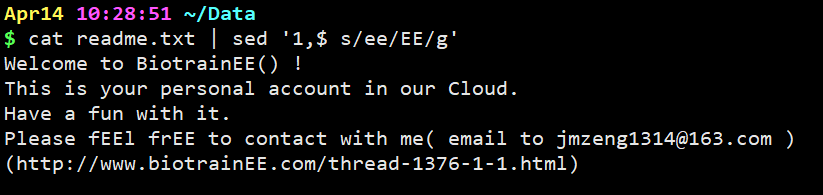
cat readme.txt | sed ‘s[a-z]\u&*g’
意思是把文本中所有小写全部转换成大写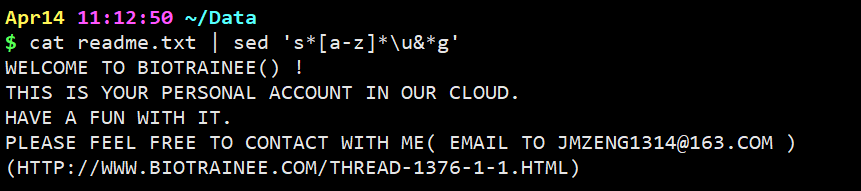
head example.gtf | awk ‘/ENSEMBL/{print $10,$12,$14}’ | sed ‘s/“//g’ | sed ‘s/;//g’![DCEWQ0OI%3~(XJYI(7]UH75.png](/uploads/projects/huluobu-fpy9r@dg3o3m/a08a7dc6372a8feffc815d2306c6b7be.png)
/pattern/ :匹配上 pattern 的行
cat readme.txt | sed ‘/www/ s/ee/EE/‘

y ∶ 转换,实现字符一对一转换,格式 ‘y/inchars/outchars/‘
cat readme.txt | sed ‘y/abcde/ABCDE/‘
意思是把文本中的所有小写的abcd替换成大写的。注意替换和被替换的字符数必须一致。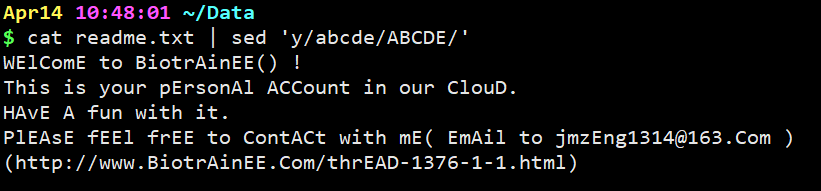
p ∶ print,把匹配或修改过的行打印出来,通常与–n参数合用
cat readme.txt | sed -n ‘/ee/p’
意思是只显示经过sed处理过的行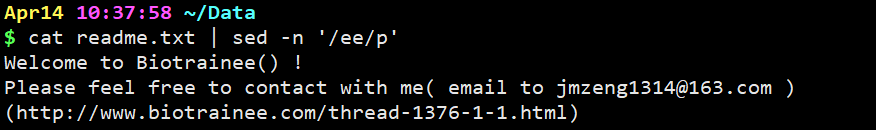

练习
3.使用head查看example.fa文件,结果传递给sed,取第2行至末尾的互补序列,并保存到新文件中
head example.fa | sed ‘2,$ y/ATCG/TAGC/‘ | cat >file8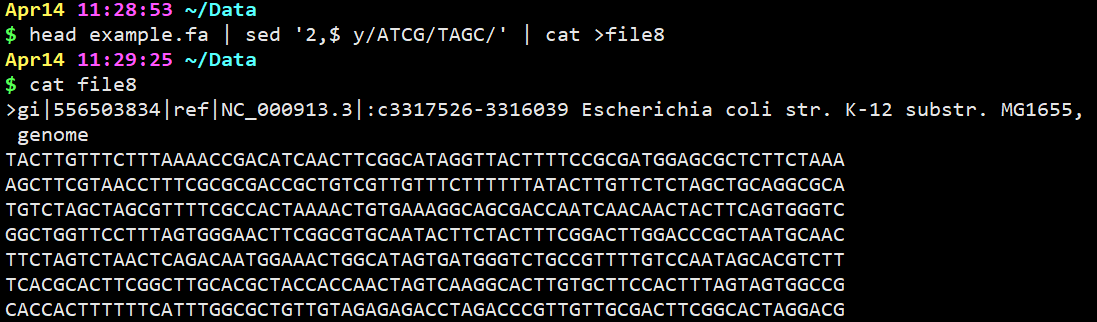
awk:
-F,fields,设置字段分隔符(默认是空白字符)
-v,var=value 定义awk程序中的一个变量及其默认值
用法:
awk [options] ‘{script}’ file
awk在读取一行文本时,会用预定义的字段分隔符划分每个数据字段,并分配给一个变量。
$0 代表整个文本行;
$1 代表文本行中的第1个数据字段;
$NF 代表文本行中的最后一个数据字段
默认的字段分隔符是任意空白字符(如:空格or 制表符),也可以用-F 参数自定义分隔符。
用less查看文本,第九列是gene_id “ENSG00000223972”类型
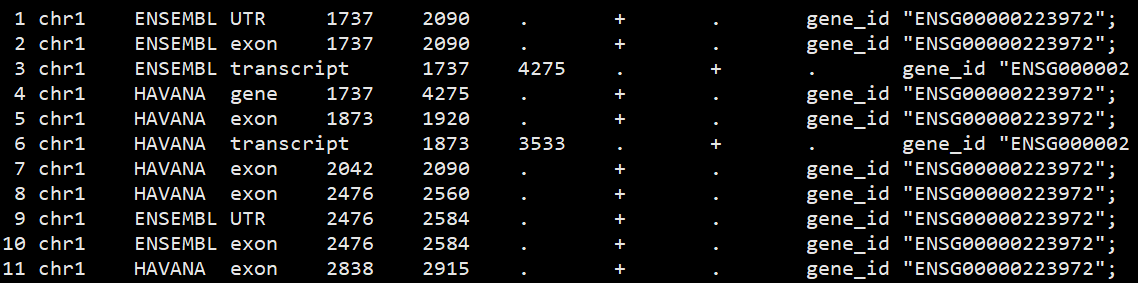
基础结构:’ {script}’
less example.gtf | awk ‘{print $9}’ | less -S
利用awk打印第九列。
(awk利用空格符为默认分隔符,所以把gene-id 与ENSG000000002223355分开了)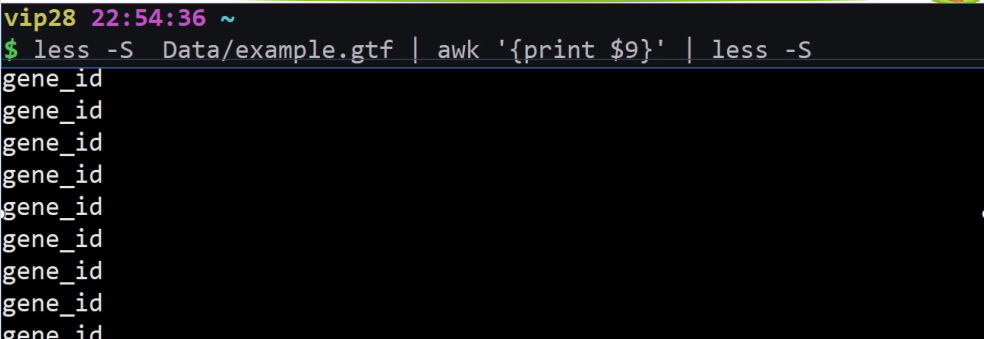
less example.gtf | awk ‘{print $9,$10}’ | less -S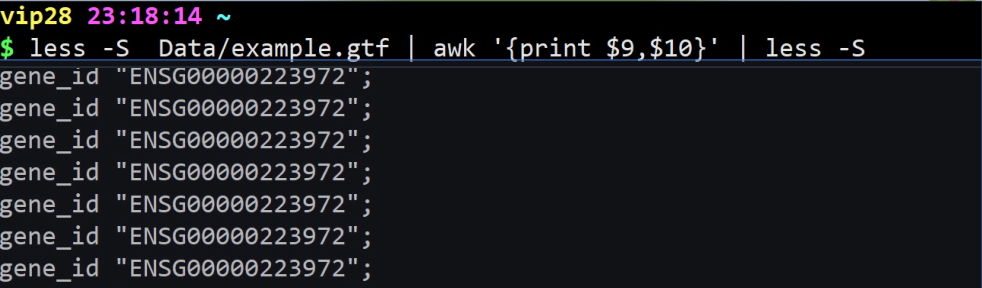
less example.gtf | awk -F ‘\t’ ‘{print $9}’ | less -S
自主设置分隔符,这里是把“\t”设置成分隔符,那么就和下面命令运行结果一样
less example.gtf | cut -f 9 | less -S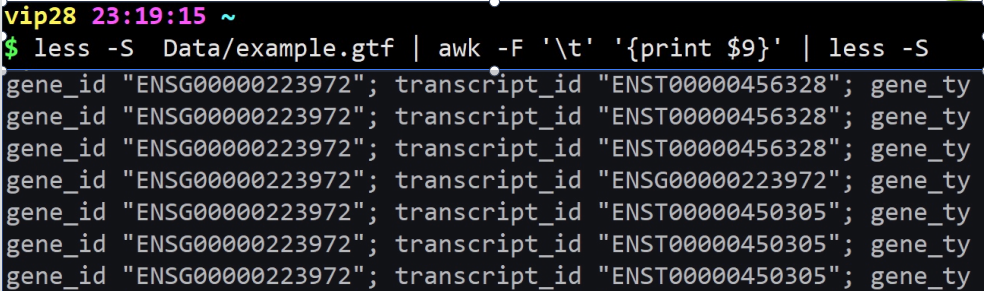
cat example.gtf | awk ‘BEGIN{FS=”\t”} {print $9}’ | head | less -S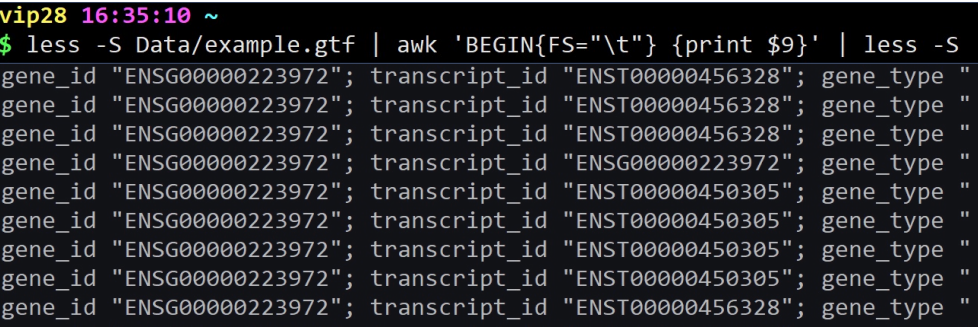
匹配结构:’ /pattern/{script}’
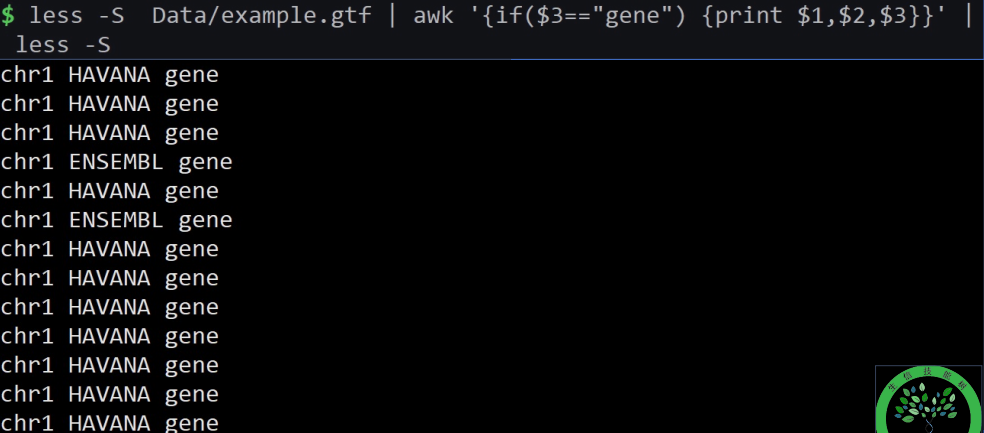
less example.gtf | awk ‘/UTR/{print $0}’ | less -S
意思是显示匹配到含有UTR的整行
$0意思是整行。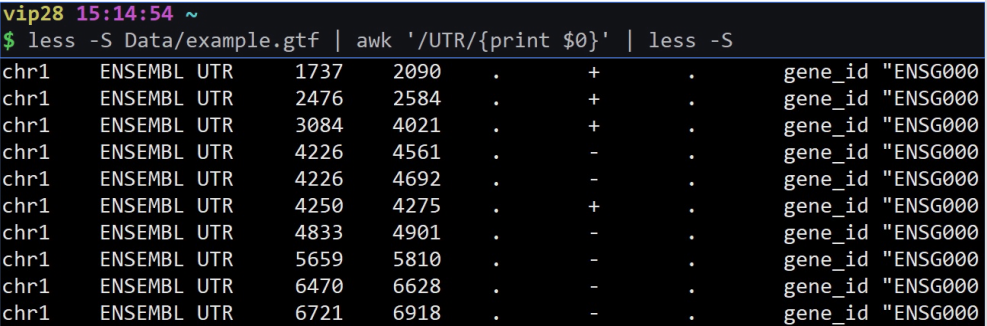
head example.gtf | awk ‘/ENSEMBL/{print $0}’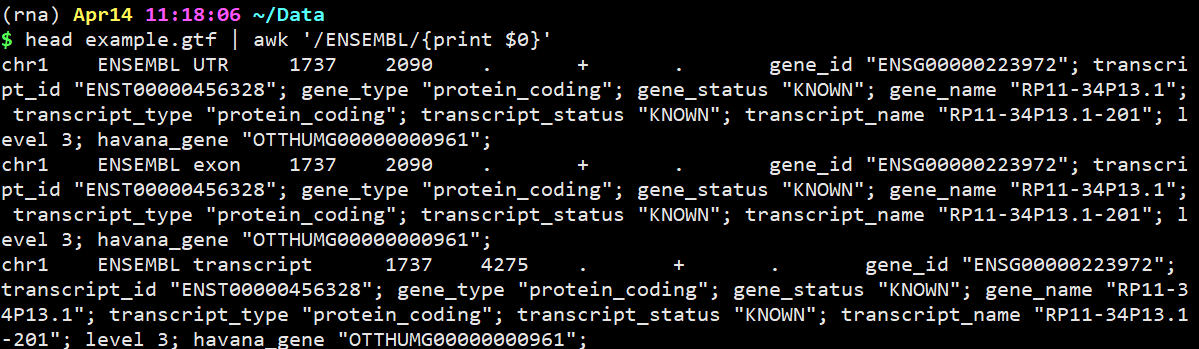
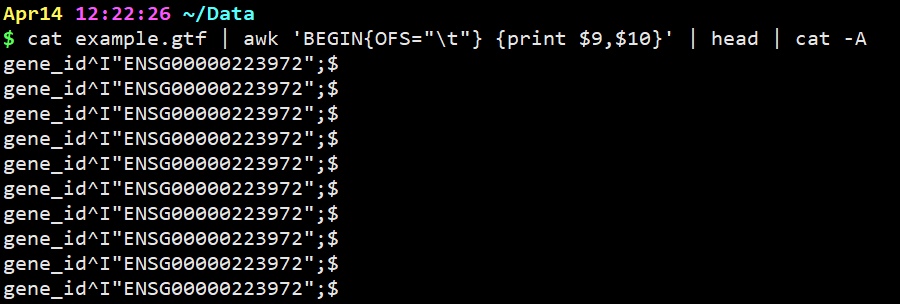
cat example.gtf | awk ‘{print $9,$10}’ | head | cat -A
意思是显示分割出来的第九列和第十列,并且显示特殊分隔符(但是因为我们在第9,10列用逗号分开,逗号在这里默认显示为空格)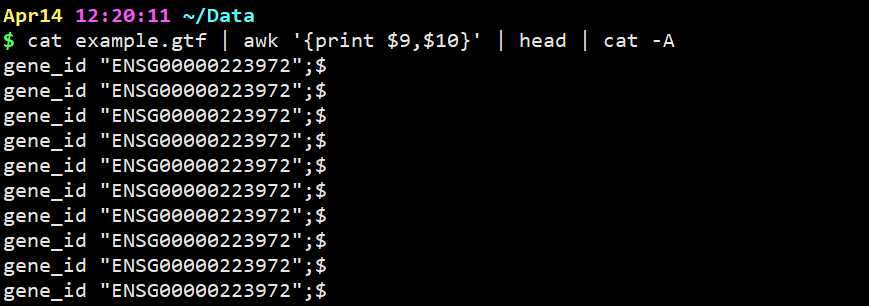 head example.gtf | awk ‘/ENSEMBL/{print $10,$12,$14}’
head example.gtf | awk ‘/ENSEMBL/{print $10,$12,$14}’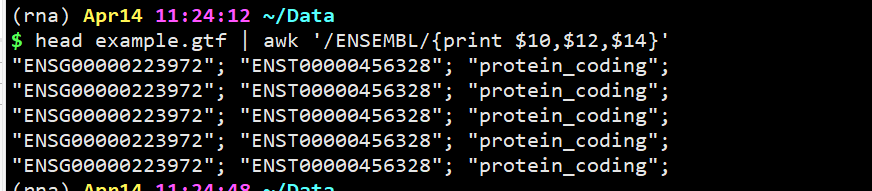
head example.gtf | awk ‘/ENSEMBL/{print $10,$12,$14}’ | tr -d ‘;”‘
head example.gtf | awk ‘/ENSEMBL/{print $10,$12,$14}’ | sed ‘s/“//g’ | sed ‘s/;//g’![F41F[UYZGD][C5~[]3)@4}Q.png](/uploads/projects/huluobu-fpy9r@dg3o3m/e661c2361ddd89b9c0f0b57c1f0f6eb9.png)
扩展结构:’BEGIN{script} {script} END{script}’
less example.gtf | awk ‘BEGIN{print “find UTR feature”} /UTR/{print $0} END{print “end”}’ | less -S
意思是在上一句代码的基础下显示匹配到UTR的整行,在开头加find UTR feature,结尾加end。
(就像日剧中男女主吃饭之前和吃饭后都会祈祷一样)
cat example.gtf | awk ‘{print $9,$10}’ | head | cat -A
意思是显示分割出来的第九列和第十列,并且显示特殊分隔符(但是因为我们在第9,10列用逗号分开,逗号在这里默认显示为空格)
cat example.gtf | awk ‘BEGIN{OFS=”\t”} {print $9,$10}’ | head | cat -A
意思是在上一步基础上,设置Tab键为分隔符并显示。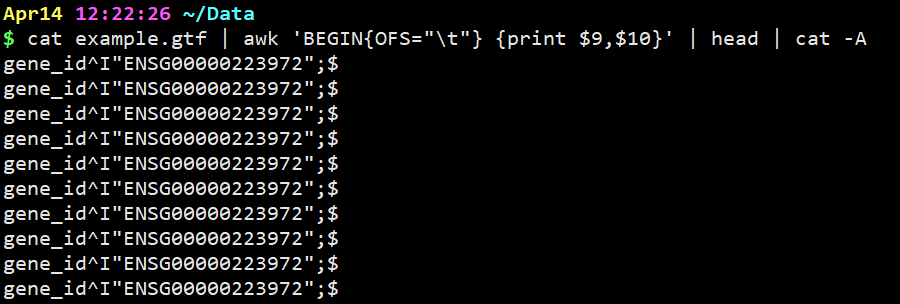
把分隔符设置成冒号试试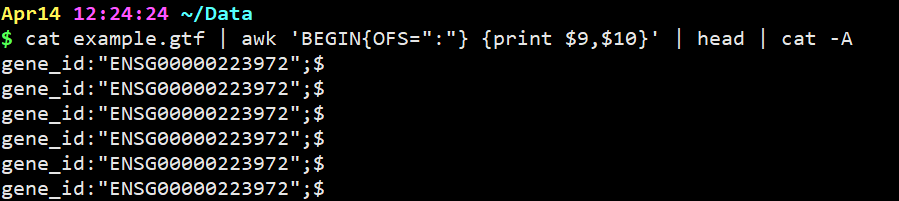
cat example.gtf | awk ‘BEGIN{FS=”\t”} {print $9}’ | head | less -S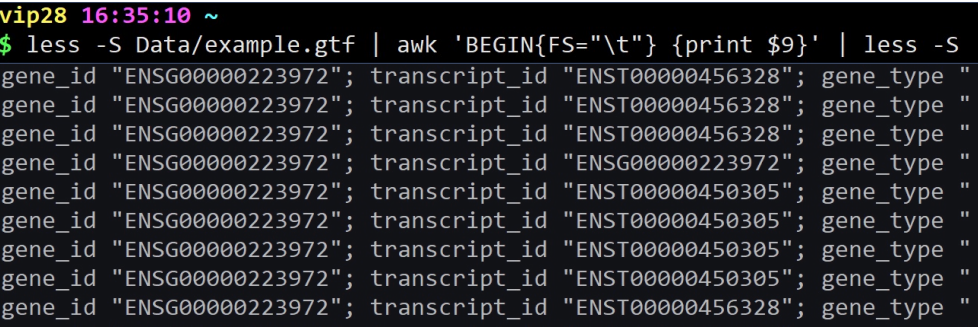
awk内置变量:
FS
OFS
NR



cat example.gtf | awk ‘BEGIN{FS=”\t”} {print NR $9}’ | head | less -S
意思是以tab键作为分隔符,打印出第九列,并且显示已处理的输入记录数,可以简单理解为行数。
awk条件和循环语句:
if:条件判断
awk ‘ { if(判断条件) {yes} else {no} } ‘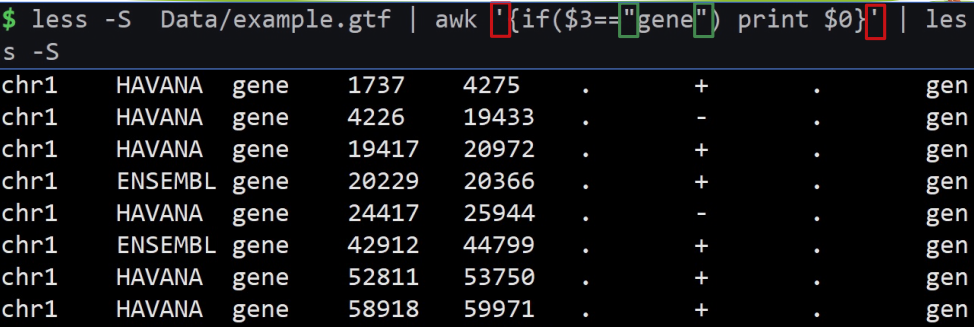

for:循环语句
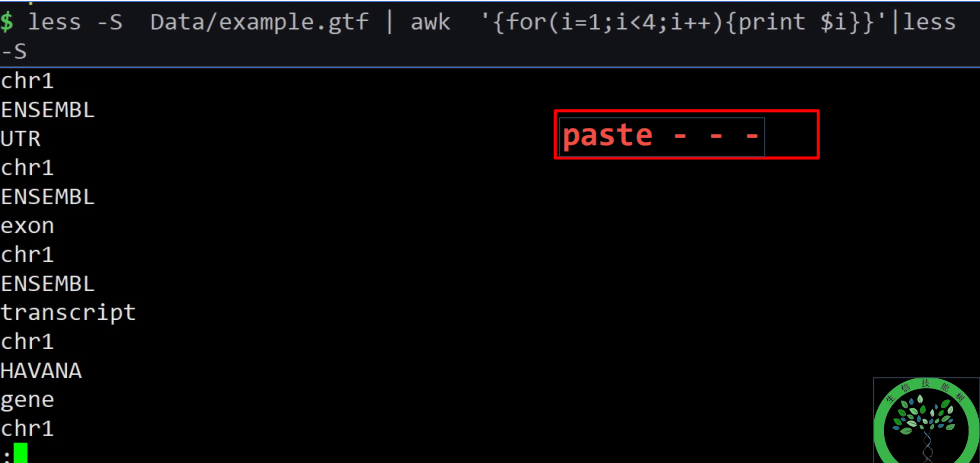
awk数学运算:
+(加),-(减),(乘),^(幂),/(除), (平方),% (取余)
int(x) x的整数部分,取靠近零一侧的值
log(x) x的自然对数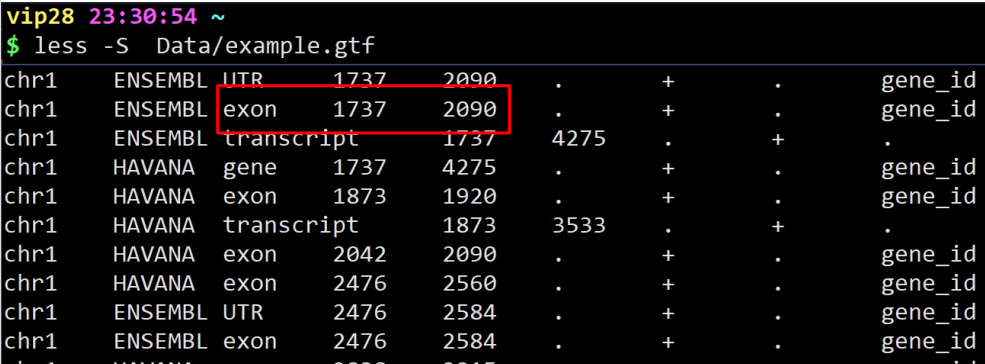
意思是匹配exon所在行,把那个且用它的第五列减去第四列*
+(加),-(减)
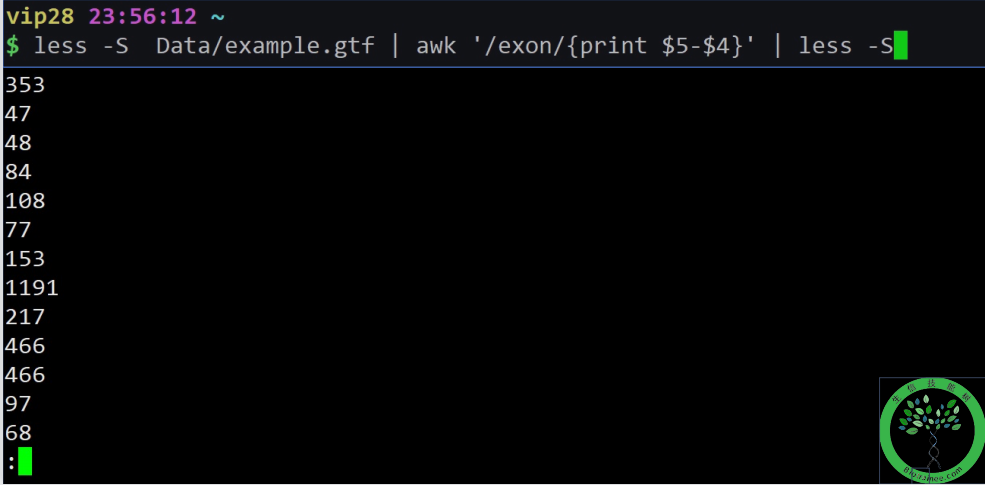
/(除)

int(x) x的整数部分,取靠近零一侧的值


若有收获,就点个赞吧
0 人点赞
