GATK 集合了一套功能全面的高通量测序数据基因组分析工具包,算是业界的权威,更新的速度非常快。需要注意的是,不同版本的 GATK 在工具应用上会有些许不同。这里我们使用是最新版本 GATK 4.1.4.1(截止2019年12月31日)。
数据准备
前面我们提到了,走GATK流程需要到官网下载很多数据库文件,比如下面这些:
dbsnp_146.hg38.vcf.gzdbsnp_146.hg38.vcf.gz.tbiMills_and_1000G_gold_standard.indels.hg38.vcf.gzMills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbiHomo_sapiens_assembly38.fastaHomo_sapiens_assembly38.fasta.gzHomo_sapiens_assembly38.fasta.faiHomo_sapiens_assembly38.dict1000G_phase1.snps.high_confidence.hg38.vcf.gz1000G_phase1.snps.high_confidence.hg38.vcf.gz.tbi
有了这些数据之后,我们就可以开始 GATK 的最佳实践流程了:
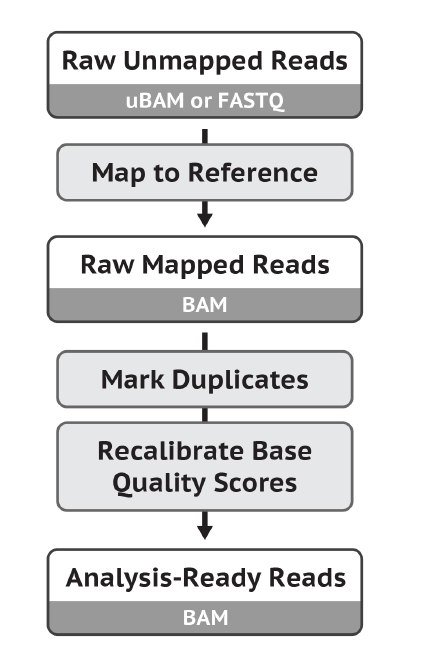
下图是官网给出的示意图,我们已经从原始的 fastq 文件经 trim 过滤后,再用 bwa 比对到参考基因组得到了 bam 文件,所以接下来走 Mark Duplicates 和 BQSR:
标记或去除重复序列
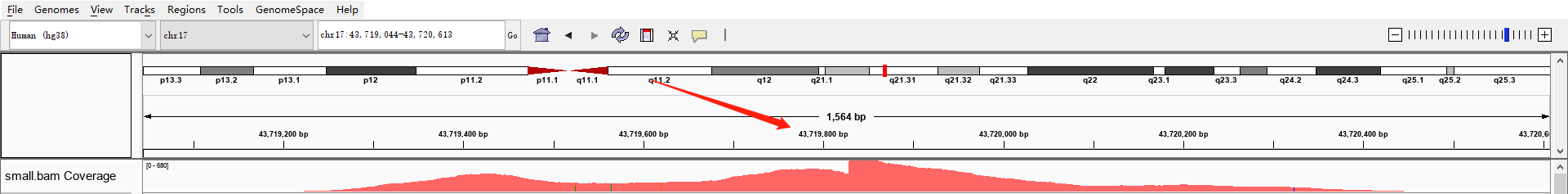
在拿到的测序结果中,有一部分序列是重复的,这是由于上机前需要进行PCR扩增,如果序列扩增次数不同,就会导致PCR重复,重复序列在igv可视化的时候就可以明显看出来。比如我们前面提取的small.bam在这个区域chr17:43,719,044-43,720,613的覆盖度就出现了断崖式的增长。
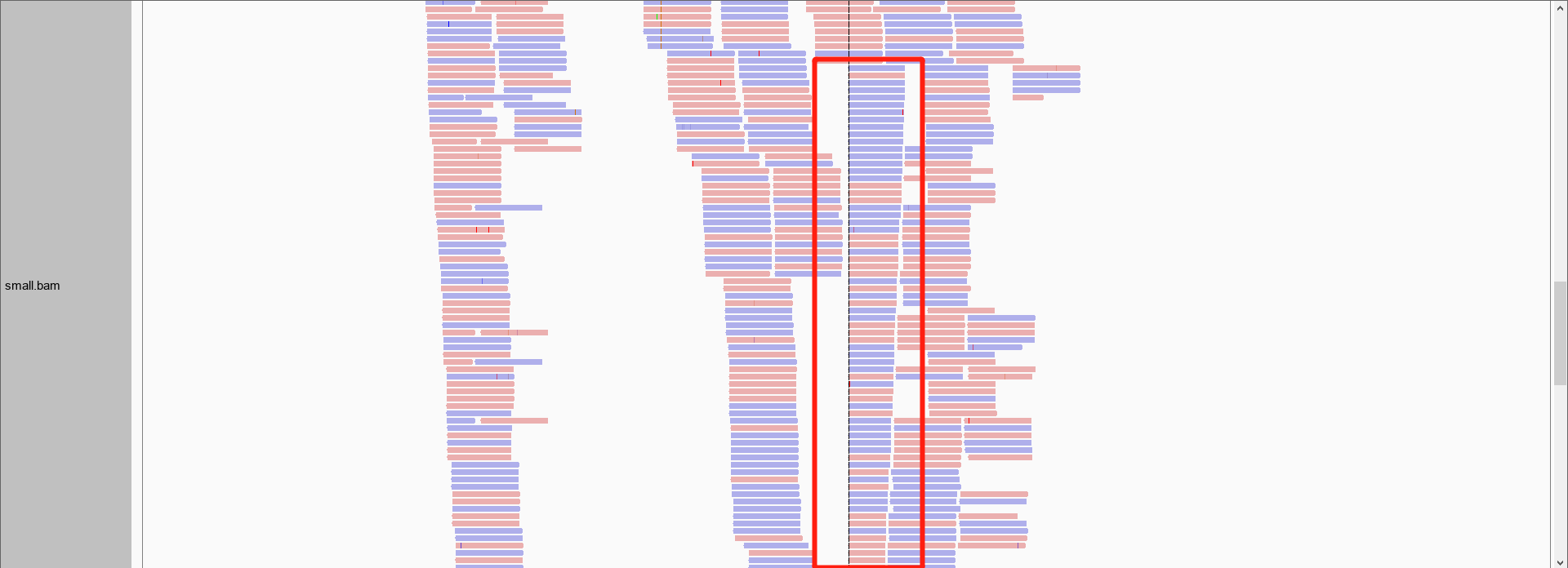
所以我们拿到比对后的bam文件之后,要进行标记重复序列甚至去除掉重复序列。用到了GATK的MarkDuplicates工具,代码如下:
GATK=~/wes_cancer/biosoft/gatk-4.1.4.1/gatkcat config | while read iddoBAM=./4.align/${id}.bamif [ ! -f ./5.gatk/ok.${id}_marked.status ]thenecho "start MarkDuplicates for ${id}" `date`$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" MarkDuplicates \-I ${BAM} \--REMOVE_DUPLICATES=true \-O ./5.gatk/${id}_marked.bam \-M ./5.gatk/${id}.metrics \1>./5.gatk/${id}_log.mark 2>&1if [ $? -eq 0 ]thentouch ./5.gatk/ok.${id}_marked.statusfiecho "end MarkDuplicates for ${id}" `date`samtools index -@ 16 -m 4G -b ./5.gatk/${id}_marked.bam ./5.gatk/${id}_marked.baifidone
解释一下上面的代码:首先把 config 文件做好,也就是我们的样本名,在这里作为一个配置文件,在前面我们也一直用它,里面记录的内容是:
$ cat configcase1_biorep_A_techrepcase1_biorep_Bcase1_biorep_C......case6_biorep_Ccase6_germlinecase6_techrep_2
进入到while read id;do...;done循环后,每 read 读取一行为一个 id,也就是一个样本名,对于每一个样本,再赋值 bam 文件的具体路径,命名为 BAM。
接下来就是一个 if 判断语句,判断是否已经存在生成的./5.gatk/ok.${id}_marked.status 文件,如果存在,就不再执行,如果不存在就继续往下走。需要注意的是,如果在 shell 命令执行后,会有一个返回值 $?,如果返回值为 0 ,只上一步命令成功执行,否则执行失败。在这里,如果命令成功执行,我就创建一个空文件 ok.${id}_marked.status。
这样写控制语句的原因,是为了避免重复提交代码。如果多个样本批量跑,跑到一半的时候任务挂了,比如服务器突然停电了,那后续再次提交命令的时候,已经完成的样本就不会被再次执行,我们要做的就是重新提交这个脚本而已。紧接着就是 echo start...date这是为了记录时间,和后面的 echo end...date相对应,这样我们可以知道跑完一个样本需要的时间,也可以在命令前加上一个 time 命令,也可以达到类似的效果。中间部分就是主要的命令:
-Xmx20G -Djava.io.tmpdir=./:设置调用的内存大小为 20G
-I $BAM:指定输入文件
--REMOVE_DUPLICATES=true:删除掉重复序列,如果不加这个参数,就只是标记重复序列而不会删除。
-O ${id}_marked.bam:输出文件
1>${id}_log.mark 2>&1:这里的1和2是捕获正确和错误输出流,把内容都输出到${id}_log.mark
最后是用samtools对生成的bam文件构建索引,这是后续步骤的要求。
简单查看一下删除重复序列的结果,去除 Duplicates 之前的总 reads 数为 212761899,删掉 Duplicates 之后剩下 192699040 了:
$ samtools view ./4.align/case1_biorep_A_techrep.bam |wc -l212761899$ samtools view ./5.gatk/case1_biorep_A_techrep_marked.bam |wc -l192699040
碱基质量重校正BQSR
因为对于 WGS 或者 WES 的测序数据,我们最关心的问题就是变异检测了,而变异检测和碱基质量值密切相关。而碱基的质量值有测序仪和测序系统产生,机器带来的系统误差和噪声是不可避免的。因此,找变异之前,需要进行碱基质量重校正BQSR(Base Quality Score Recalibration)。用到了GATK的两个工具BaseRecalibrator和ApplyBQSR,代码如下:
GATK=~/wes_cancer/biosoft/gatk-4.1.4.1/gatksnp=~/wes_cancer/data/dbsnp_146.hg38.vcf.gzindel=~/wes_cancer/data/Mills_and_1000G_gold_standard.indels.hg38.vcf.gzref=~/wes_cancer/data/Homo_sapiens_assembly38.fastacat config | while read iddoif [ ! -f ./5.gatk/${id}_bqsr.bam ]thenecho "start BQSR for ${id}" `date`$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" BaseRecalibrator \-R $ref \-I ./5.gatk/${id}_marked.bam \--known-sites ${snp} \--known-sites ${indel} \-O ./5.gatk/${id}_recal.table \1>./5.gatk/${id}_log.recal 2>&1$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" ApplyBQSR \-R $ref \-I ./5.gatk/${id}_marked.bam \-bqsr ./5.gatk/${id}_recal.table \-O ./5.gatk/${id}_bqsr.bam \1>./5.gatk/${id}_log.ApplyBQSR 2>&1echo "end BQSR for ${id}" `date`fidone
关于上面的两个工具的解释:
第一个,BaseRecalibrator,这里计算出了所有需要进行重校正的reads和特征值,然后把这些信息输出为一份校准表文件(recal.table)。--known-sites参数需要输入已知且可靠的变异位点,可以有多个。因为人与人之间基因组的差异其实是很小的,约 0.1%,那么在一个群体中发现的已知变异,在某个人身上也很可能是同样存在的。所以,我们首先排除掉所有的已知变异位点,然后计算每个(报告出来的)质量值下面有多少个碱基在比对之后与参考基因组上的碱基是不同的,这些不同碱基就被我们认为是错误的碱基,它们的数目比例反映的就是真实的碱基错误率。
第二个,ApplyBQSR,这一步利用第一步得到的校准表文件(recal.table)重新调整原来BAM文件中的碱基质量值,并使用这个新的质量值重新输出一份新的BAM文件。
校正前后的碱基质量值:
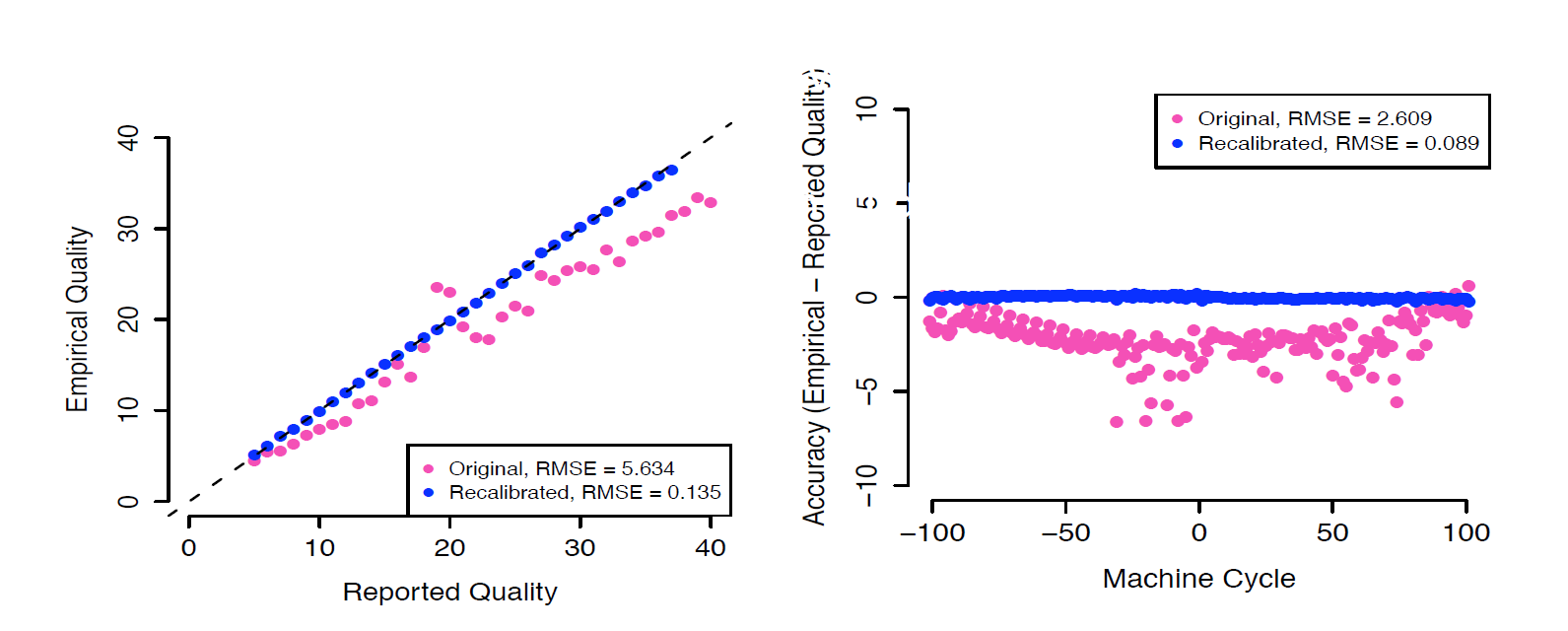
单样本找 Germline SNPS + INDELs
我们通常说的突变,可以分为种系突变 Germline mutations(又称生殖突变)和体细胞突变 Somatic mutations。前者是指在生殖细胞中发生的任何可检测、可遗传的突变。在生殖细胞系以外的细胞中发生的突变称为体细胞突变,也称作获得性突变,是不可遗传的。使用 GATK 的流程进行 Germline mutations,主要用到的工具是 HaplotypeCaller,流程示意图如下(最后得到的 vcf 文件应该还有走 VQSR 进行过滤):
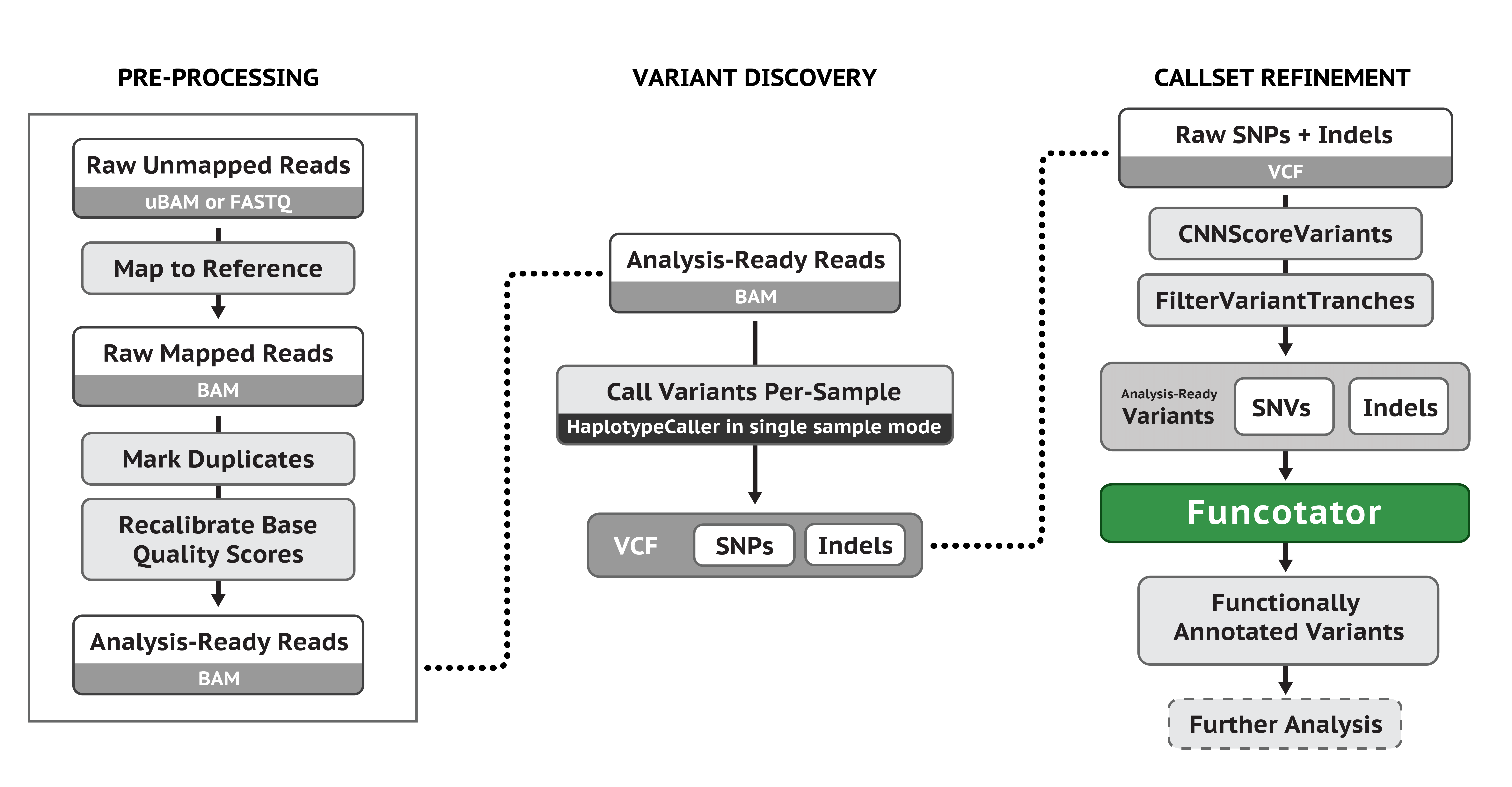
在这里我使用到的脚本是:
GATK=~/wes_cancer/biosoft/gatk-4.1.4.1/gatksnp=~/wes_cancer/data/dbsnp_146.hg38.vcf.gzindel=~/wes_cancer/data/Mills_and_1000G_gold_standard.indels.hg38.vcf.gzref=~/wes_cancer/data/Homo_sapiens_assembly38.fastabed=~/wes_cancer/data/hg38.exon.bedcat config | while read iddoecho "start HC for ${id}" `date`$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" HaplotypeCaller -ERC GVCF \-R ${ref} \-I ./5.gatk/${id}_bqsr.bam \--dbsnp ${snp} \-L ${bed} \-O ./5.gatk/${id}_raw.vcf \1>./5.gatk/${id}_log.HC 2>&1echo "end HC for ${id}" `date`donecd ./5.gatk/gvcffor chr in chr{1..22} chrX chrY chrMdotime $GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" GenomicsDBImport \-R ${ref} \$(ls ./*raw.vcf | awk '{print "-V "$0" "}') \-L ${chr} \--genomicsdb-workspace-path gvcfs_${chr}.dbtime $GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" GenotypeGVCFs \-R ${ref} \-V gendb://gvcfs_${chr}.db \-O gvcfs_${chr}.vcfdone$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" GatherVcfs \$(for i in {1..22} X Y M;do echo "-I gvcfs_chr${i}.vcf" ;done) \-O merge.vcf
mutect 找 Somatic mutations
前面单样本走 GATK HaplotypeCaller 流程得到 vcf 文件主要包含了 germline mutations 信息。但是我们这里分析的是肿瘤样本,包含成对的 tumor 和 normal 样本,更加关心的是 tumor 样本中所包含的 somatic mutations 信息。所以我们应该走 GATK 的 somatic mutations 流程:
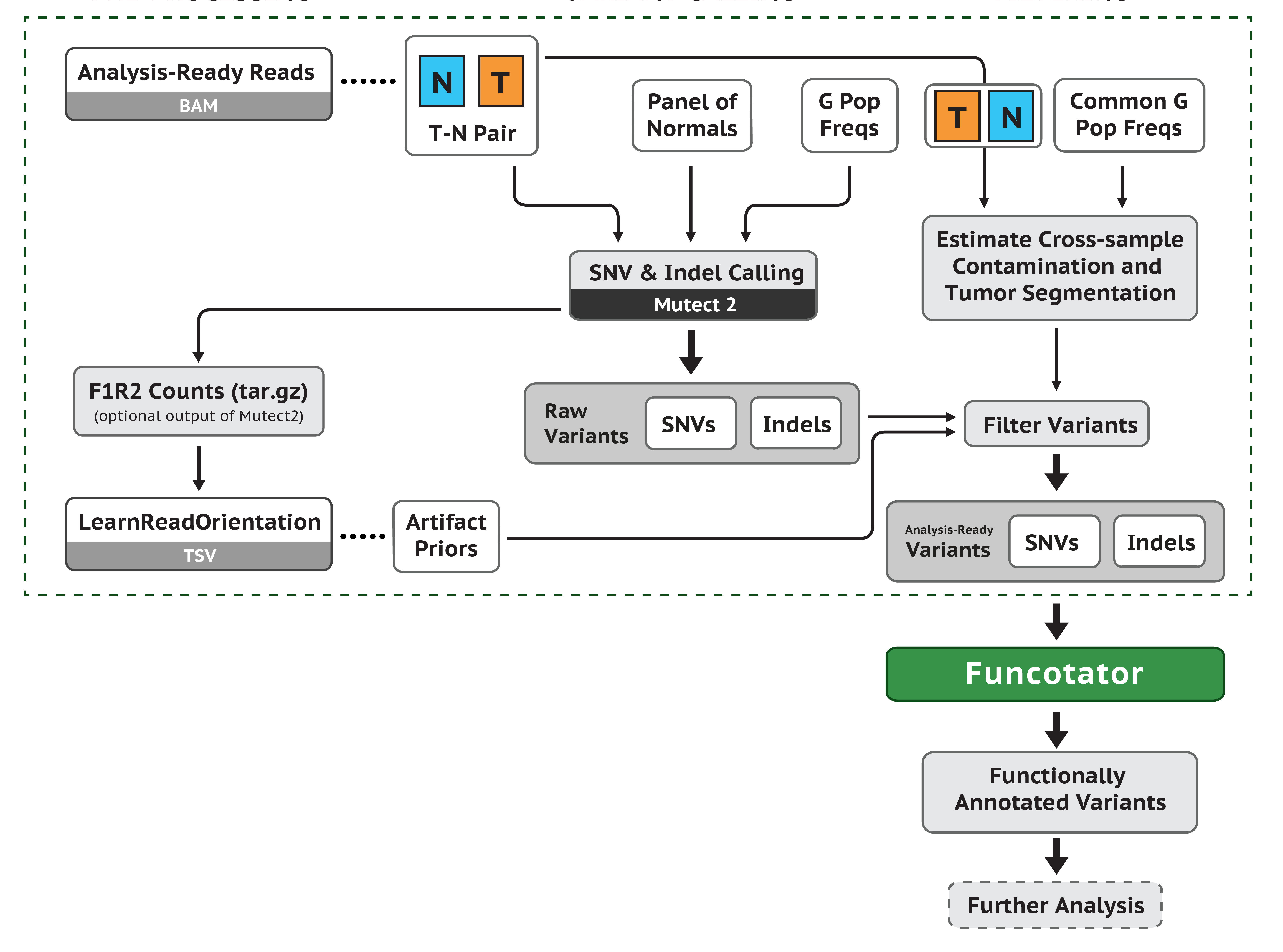
我们这里有成对的 normal 和 tumor样本,需要把配置文件 config2 做成两列,如下:
$ cat config2case1_germline case1_biorep_A_techrepcase2_germline case2_biorep_Acase3_germline case3_biorep_A......case4_germline case4_techrep_2case5_germline case5_techrep_2case6_germline case6_techrep_2
详细代码如下:
## mutect.shGATK=~/wes_cancer/biosoft/gatk-4.1.4.1/gatkref=~/wes_cancer/data/Homo_sapiens_assembly38.fastabed=~/wes_cancer/data/hg38.exon.bedcat config2 | while read iddoarr=(${id})sample=${arr[1]}T=./5.gatk/${arr[1]}_bqsr.bamN=./5.gatk/${arr[0]}_bqsr.bamecho "start Mutect2 for ${id}" `date`$GATK --java-options "-Xmx20G -Djava.io.tmpdir=./" Mutect2 -R ${ref} \-I ${T} -tumor $(basename "$T" _bqsr.bam) \-I ${N} -normal $(basename "$N" _bqsr.bam) \-L ${bed} \-O ./6.mutect/${sample}_mutect2.vcf$GATK FilterMutectCalls \-R ${ref} \-V ./6.mutect/${sample}_mutect2.vcf \-O ./6.mutect/${sample}_somatic.vcfecho "end Mutect2 for ${id}" `date`cat ./6.mutect/${sample}_somatic.vcf | perl -alne '{if(/^#/){print}else{next unless $F[6] eq "PASS";next if $F[0] =~/_/;print } }' > ./6.mutect/${sample}_filter.vcfdone
关于上面代码的解释:首先是Mutect2,主要是根据对正常-肿瘤样本进行位点比较寻找体细胞突变somatic mutations。如果没有正常样本,也可以使用mutect2软件,流程需要进行修改,而且结果的假阳性会很高。我们这里输入了两个配对的 T-N 样本 -I ${T} -tumor $(basename "$T" _bqsr.bam)和-I ${N} -normal $(basename "$N" _bqsr.bam)。这样我们就拿到了每个 tumor 样本中的突变位点,${sample}_mutect2.vcf。但是这还不够,还要通过FilterMutectCalls过滤,过滤后就是可信度较高的somatic突变位点会被打上PASS标签,就是${sample}_somatic.vcf。最后我们再用一个简单的脚本把打上PASS标签的位点提取出来,得到了${sample}_filter.vcf,如:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT case1_biorep>chr1 6146376 . G T . PASS DP=184;ECNT=1;NLOD=22.56;N_A>chr1 6461445 . G T . PASS DP=21;ECNT=1;NLOD=2.70;N_ART>chr1 31756671 . C A . PASS DP=172;ECNT=1;NLOD=9>chr1 32672798 . A T . PASS DP=448;ECNT=1;NLOD=2>chr1 39441098 . G T . PASS DP=411;ECNT=1;NLOD=3>......
到这里拿到了最后的 vcf 文件,我们就可以进一步分析,而GATK的流程也到这里差不多结束,希望你也能学会这个分析流程。

若有收获,就点个赞吧
0 人点赞
