在前面两节中,我们比较了不同 vcf 注释软件的使用,以及将注释后的记过转换成了 maf 格式的文件,由于snpeff 注释结果暂时无法转换成 maf,所以后面我们就比较 ANNOVAR、VEP、GATK Funcatator 的注释结果得到的合并后的 maf 文件。首先将拿到的结果载入到R语言中,然后用 maftool 可视化,代码如下:
读入数据
rm(list = ls())require(maftools)options(stringsAsFactors = F)## annovarannovar.laml <- annovarToMaf(annovar = "./7.annotation/annovar/annovar_merge.vcf",refBuild = 'hg38',tsbCol = 'Tumor_Sample_Barcode',table = 'refGene',MAFobj = T)## gatk# gatk.laml = read.maf(maf = 'gatk/gatk4.1.4.0_merge.maf')library(data.table)tmp=fread('./7.annotation/funcatator/funcatator_merge.maf')gatk.laml = read.maf(maf = tmp)## vepvep.laml = read.maf(maf = './7.annotation/vep/vep_merge.maf')## for vep.lamllibrary(stringr)vep.laml@data$Protein_Change = paste0("p.",str_sub(vep.laml@data$Amino_acids,1,1),vep.laml@data$Protein_position,str_sub(vep.laml@data$Amino_acids,3,3))## save Rdatasave(annovar.laml, gatk.laml, vep.laml, file = 'laml.Rdata')## Summarylaml=annovar.lamlunique(laml@data$Tumor_Sample_Barcode)getSampleSummary(laml)getGeneSummary(laml)getFields(laml)laml=gatk.lamlunique(laml@data$Tumor_Sample_Barcode)getSampleSummary(laml)getGeneSummary(laml)getFields(laml)laml=vep.lamlunique(laml@data$Tumor_Sample_Barcode)getSampleSummary(laml)getGeneSummary(laml)getFields(laml)
mafSummary
上述步骤是把拿到的 maf 文件读入 R 中,初步处理后保存成 Rdata,并且对数据进行初步了解。接下来是绘图可视化 mafSummary
rm(list = ls())require(maftools)options(stringsAsFactors = F)load(file = 'laml.Rdata')laml=c(annovar.laml,gatk.laml,vep.laml)## mafsummaryanno=c('annovar','gatk','vep')for (i in 1:3) {#i = 1png(paste0('plotmafSummary_', anno[i], '.png'),res = 150,width = 1080,height = 1080)plotmafSummary(maf = laml[[i]],rmOutlier = TRUE,showBarcodes = T,textSize = 0.4,addStat = 'median',dashboard = TRUE,titvRaw = FALSE)dev.off()}
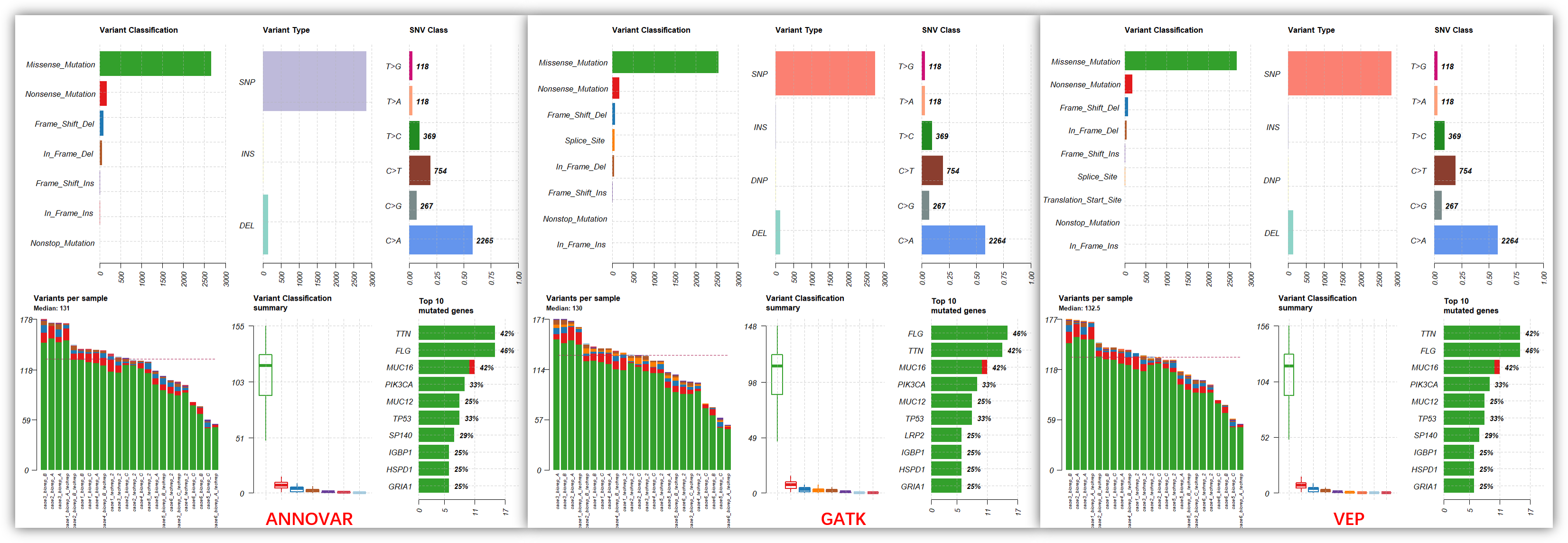
可以看到,3 个软件的注释结果总体上非常接近的,但是还是有一些小细节不同,比如我们看一下,top30 基因的oncoplot
oncoplot
## oncoplot_top30for (i in 1:3) {#i = 1png(paste0('oncoplot_top30_', anno[i], '.png'),res = 150,width = 1080,height = 1080)oncoplot(maf = laml[[i]],top = 30,fontSize = 0.5,sampleOrder = laml[[i]]@clinical.data$Tumor_Sample_Barcode,showTumorSampleBarcodes = T)dev.off()}
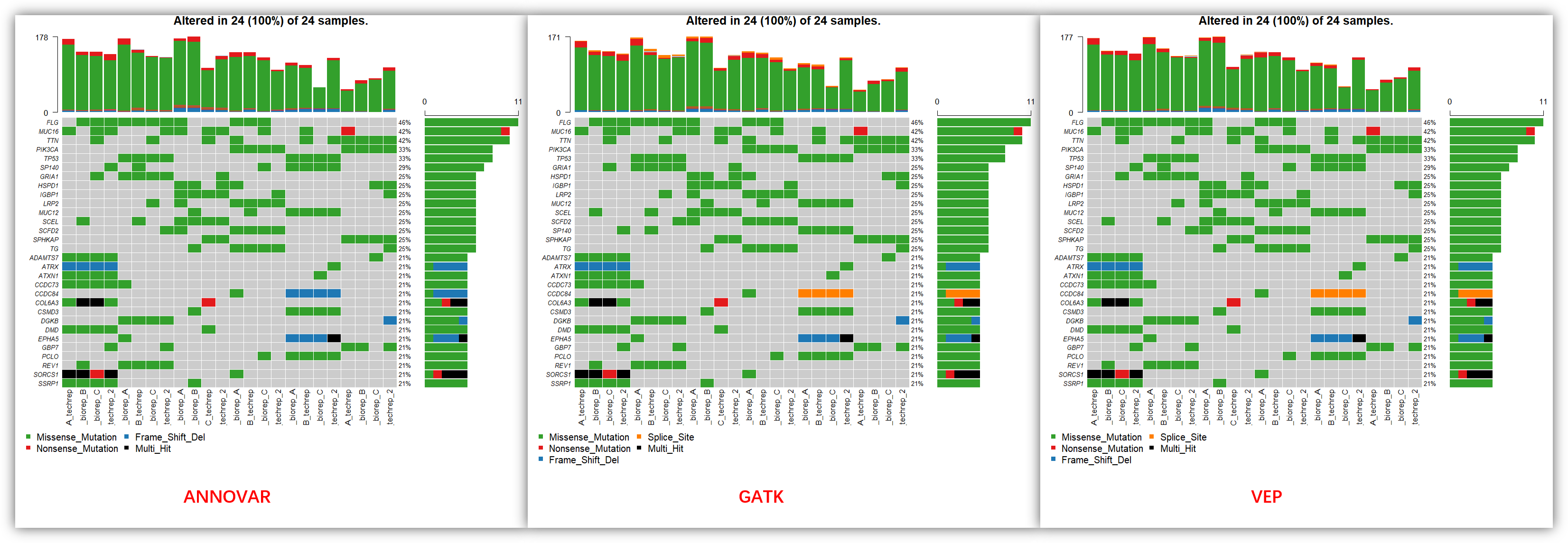
从上面的图可以看到,3 个软件注释后的可视化结果非常接近,当然也不是完全一模一样,比如说 SP140 基因,在 ANNOVAR 和 VEP 的注释结果都是排在了第 5 位,而在 GATK 的注释结果就排到了第 13 位,少了一个突变位点。
lollipopPlot
下面通过 lollipopPlot 函数来进一步比较一下:
## lollipopPlot for SP140gene='SP140'protein=c("AAChange.refGene","Protein_Change","Protein_Change")for (i in 1:3) {#i=3png(paste0(gene,'_', anno[i], '.png'),res = 150,width = 1080,height = 1080)maftools::lollipopPlot(maf = laml[[i]],gene = gene,AACol = protein[i],labelPos = 'all')dev.off()}
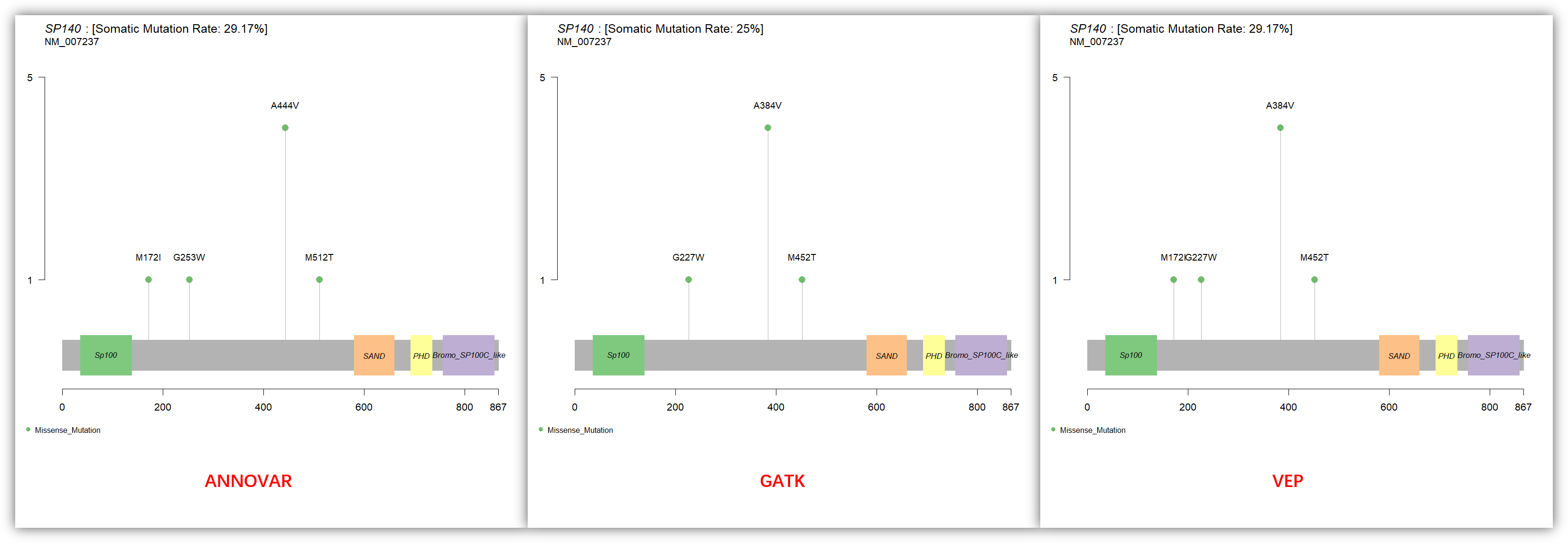
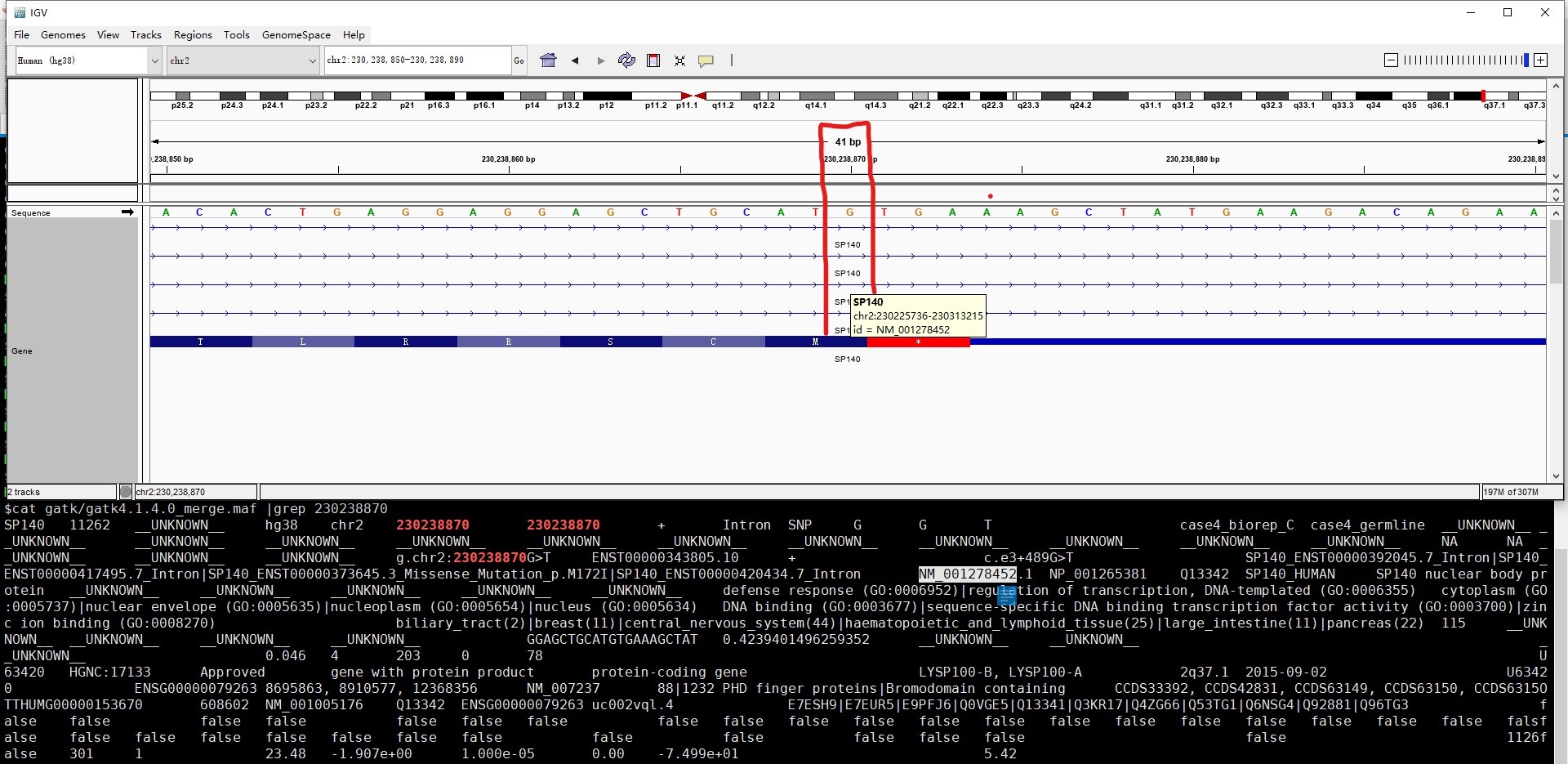
可以看到 GATK 比另外两个软件的注释结果少了一个位点,且三个软件的氨基酸的坐标也不完全一致。我猜是注释到不同转录本的原因,结合 IGV 进行查看一下,果然如此,GATK 选择的转录本是 NM_001278452.1,在 IGV 中这个转录本该位点 230238870 为内含子,同样导致氨基酸坐标不一致也是因为注释到不同的转录本:
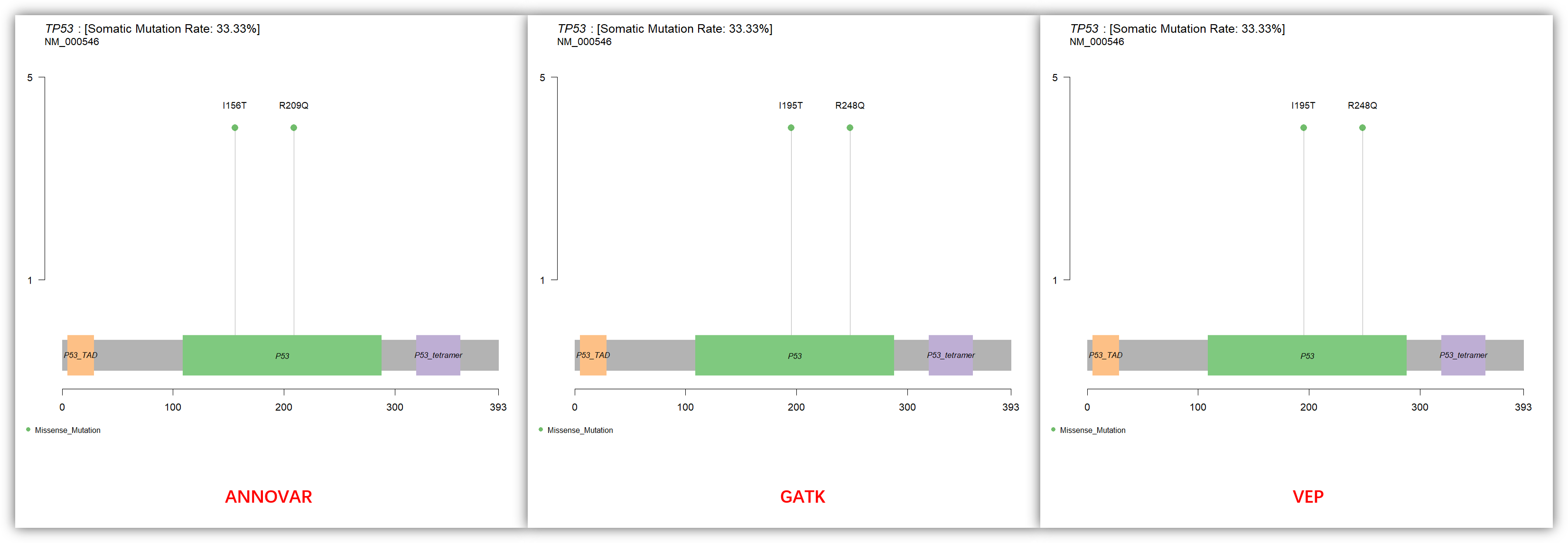
最后我们再来比较 TP53 基因的注释结果,也是因为转录本的不同导致氨基酸坐标不一致:
## lollipopPlot for TP53gene='TP53'protein=c("AAChange.refGene","Protein_Change","Protein_Change")for (i in 1:3) {#i=3png(paste0(gene,'_', anno[i], '.png'),res = 150,width = 1080,height = 1080)maftools::lollipopPlot(maf = laml[[i]],gene = gene,AACol = protein[i],labelPos = 'all')dev.off()}
maftools 还可以绘制很多图,完成更多的分析。感兴趣的朋友可以试试。

若有收获,就点个赞吧
0 人点赞
