其实是补一下再重温一下最近老师讲课了,知识是跟的上的,但是一些具体的内容就有些乏力了,然后想了想还是视觉的一些命令的参数不记得这方面影响更大一些
以及这里我会把前面学习到的一些命令参数在这理手动打一下,用以重温
基础知识
fastq的数据基本知识

以及,每一段都是不同的身份标识符,使用冒号进行分割 最早的是测序仪的名字 
 为了避免空值,会Q+33或Q+64,如果是后者的话,ASCII码会出现小写字母
为了避免空值,会Q+33或Q+64,如果是后者的话,ASCII码会出现小写字母
Q20 指的是碱基质量值大于20的比例,后面同理。肯定越高越好,大约85以上已经很好了
转录分析流程

课后习题
 (在后面我讨论)
(在后面我讨论)1.统计reads_1.fq文件种共有多少条reads?
# 答案不只一种,看看你能用集中方法算出来# NR表示行号# %表示除于取整、zless SRR1039510_1.fastq.gz | grep '^@SRR' |wc -lzless -S SRR1039510_1.fastq.gz | paste - - - - |wc -lzless SRR1039510_1.fastq.gz |wc -l | awk '{print $0/4}'zless -S SRR1039510_1.fastq.gz |awk '{ if(NR%4==2) {print} }' |wc -l# sed 版本 课后习题
2.输出reads_1.fq文件中所有的序列ID(即第一行)
zless SRR1039510_1.fastq.gz | grep '^@SRR' |less -Szless SRR1039510_1.fastq.gz | paste - - - - |cut -f 1 |less -Szless -S SRR1039510_1.fastq.gz |awk '{if(NR%4==1){print}}' |less -S
3.输出SRR1039510_1.fastq.gz文件中所有的序列(即第二行)
zless SRR1039510_1.fastq.gz | paste - - - - |cut -f 2 |less -Szless -S SRR1039510_1.fastq.gz |awk '{if(NR%4==2){print}}' |less -S
4.统计SRR1039510_1.fastq.gz碱基总数
# 简单版本zless -S SRR1039510_1.fastq.gz |paste - - - - |cut -f 2 |tr -d '\n' |wc -mzless -S SRR1039510_1.fastq.gz |paste - - - - |cut -f 2 |grep -o [ATCGN] |wc -l# awk的高阶用法:BEGIN END模块zless -S SRR1039510_1.fastq.gz |awk '{ if(NR%4==2){print} }' | awk 'BEGIN {num=0} {num=num+length($0)} END{ print "num="num}'
这里我补充部分笔记<br />针对于第一题<br />首先reads 是一条,碱基是一个,都是第二段的部分<br />首先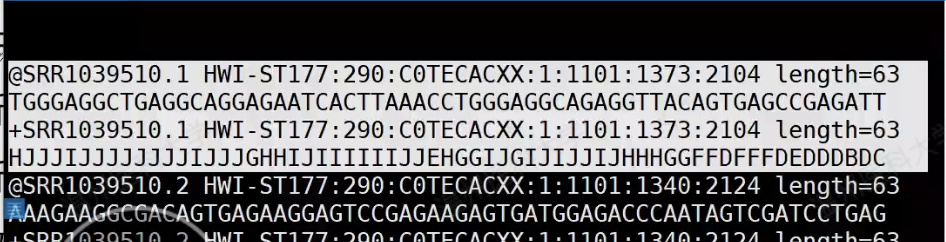,四行为一个单位,要的是第二行。<br />文件是一个gz文件,用zless解析,在用wc -l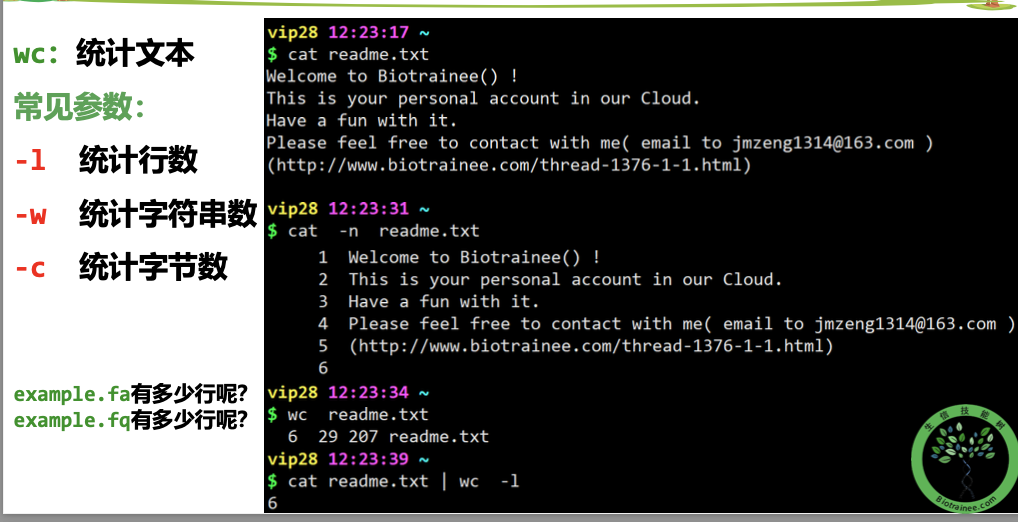的结果**除以4 **<br />**(但是值得注意的是,以上是练习数据,所以比较短,会很快得出结果,真实的数据会很大,单靠这段会卡 这里可以用ls -h看一下文件大小)h:以人类可读的方式显现容量**<br />还有一种方式**paste - - - -,意思是讲每四行转换为一行** 再wc -l,但是这里值得注意的是,最后输出显示用less -S,因为前面处理之后就不是压缩文件了如果想要使用答案中的grep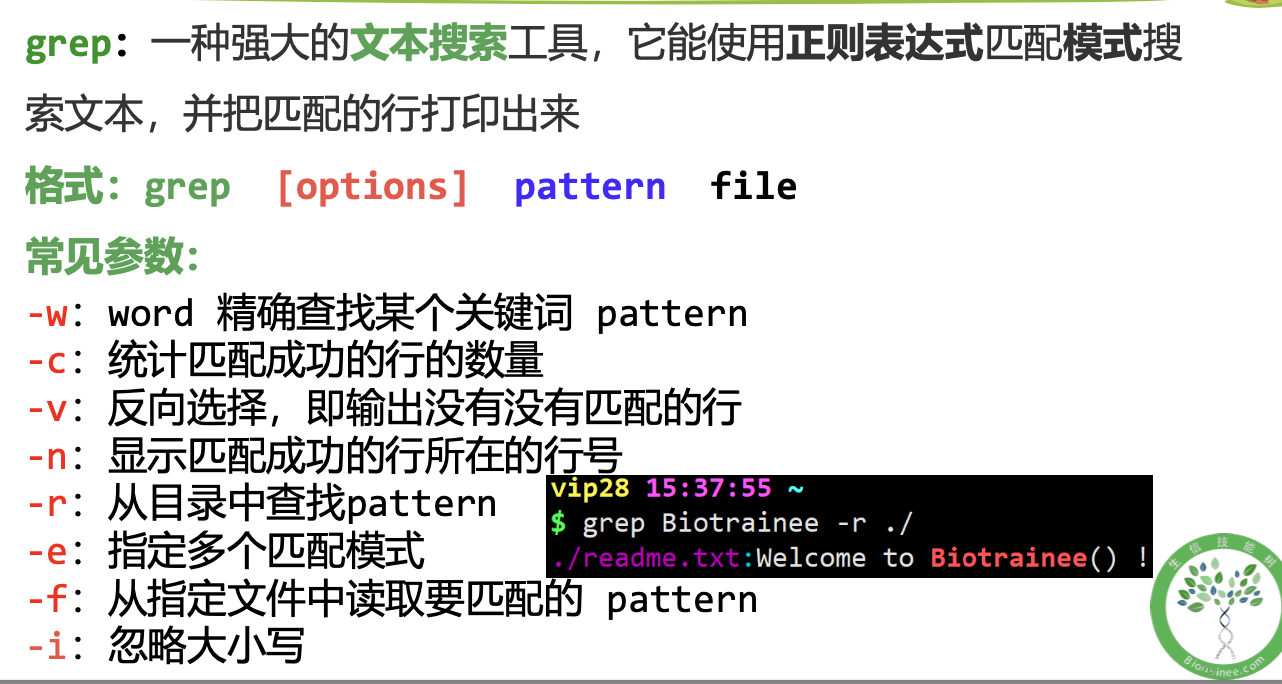的方法,这样直接输入'^@'是不对的,因为后面碱基质量值有可能是以@符号开头的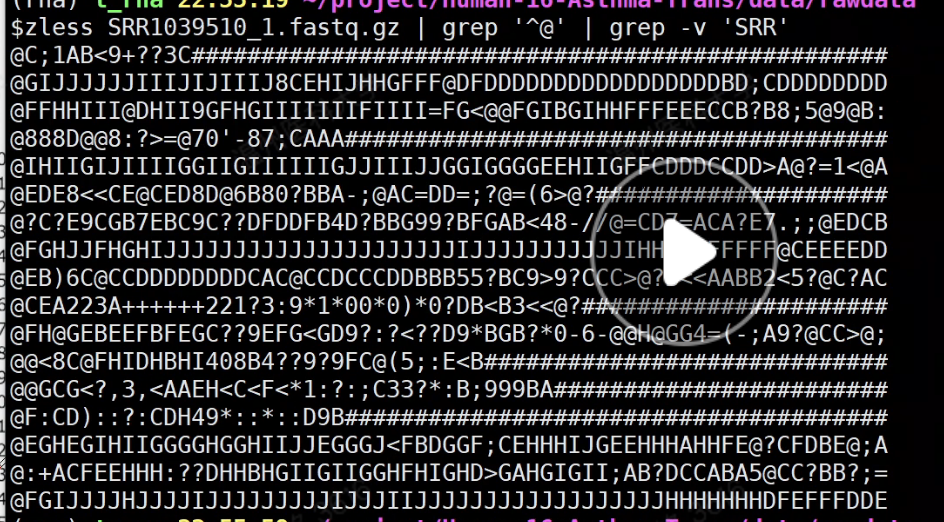(这里看一眼)这里还有awk版本的答案 (NR表示行号)<br />zless -S SRR1039510_1.fastq.gz |awk '{ if(NR%4==2) {print} }' |wc -l 其实就是要的是第二行,做个除法<a name="MP2MZ"></a>## 使用fastqc对原始数据进行质量评估<a name="LXImV"></a>### 基本概念<a name="LfQVV"></a>### 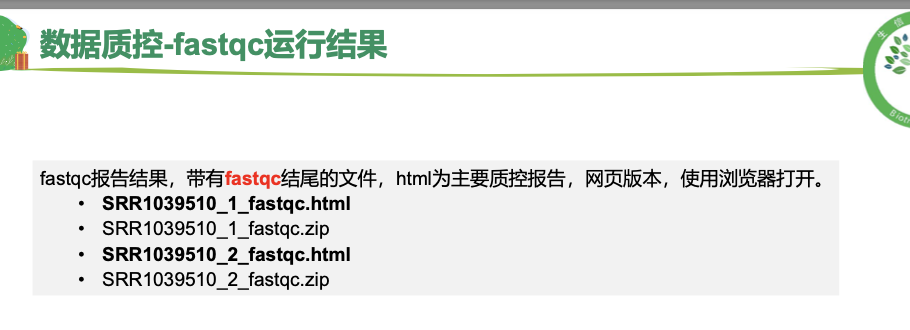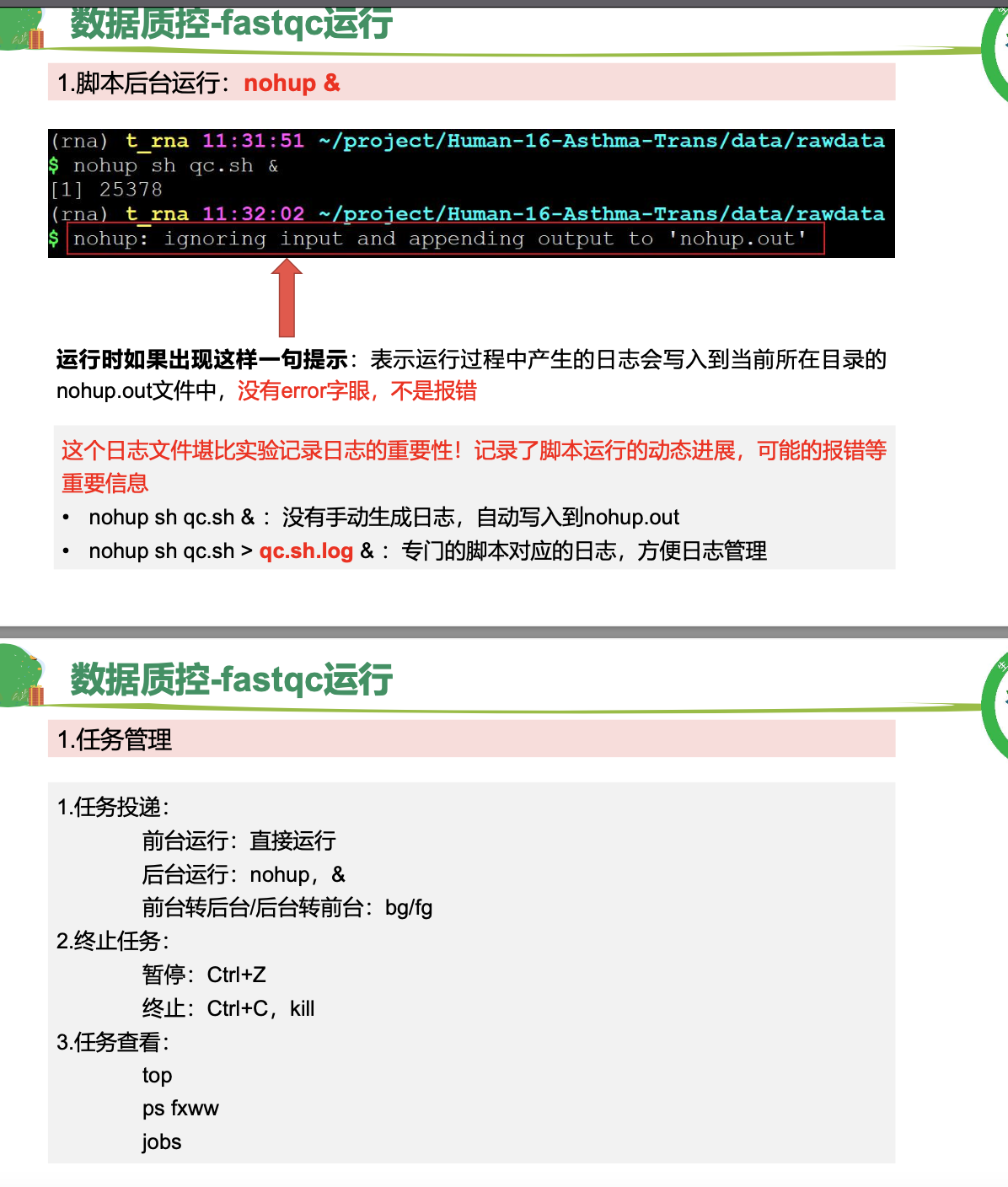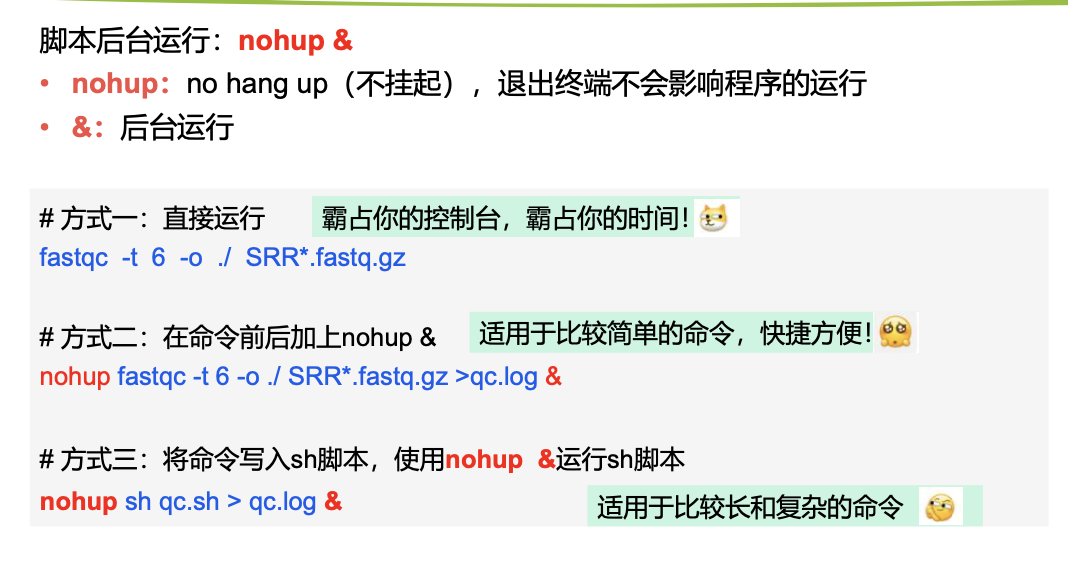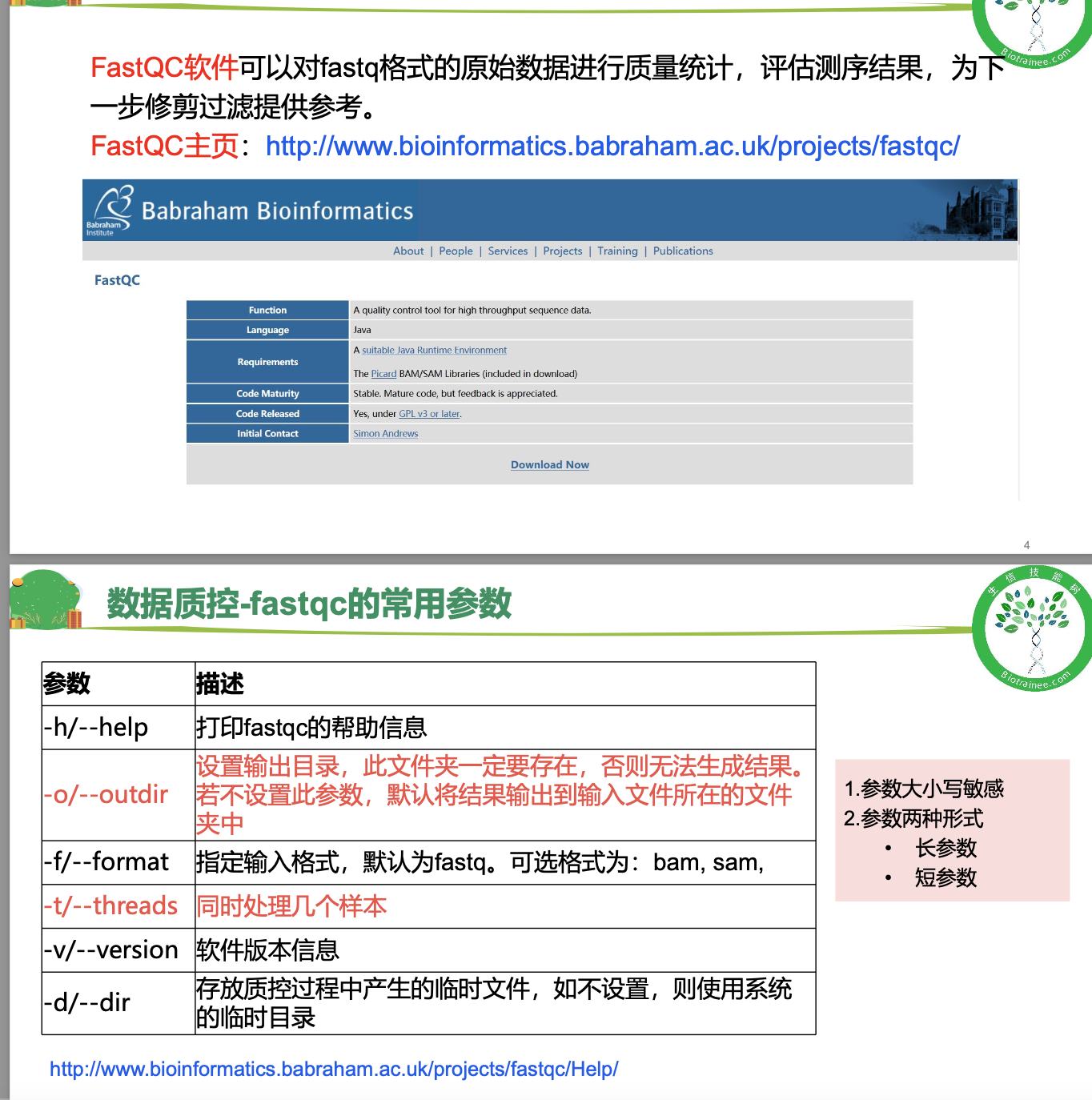<a name="c1Z9W"></a>### 数据解读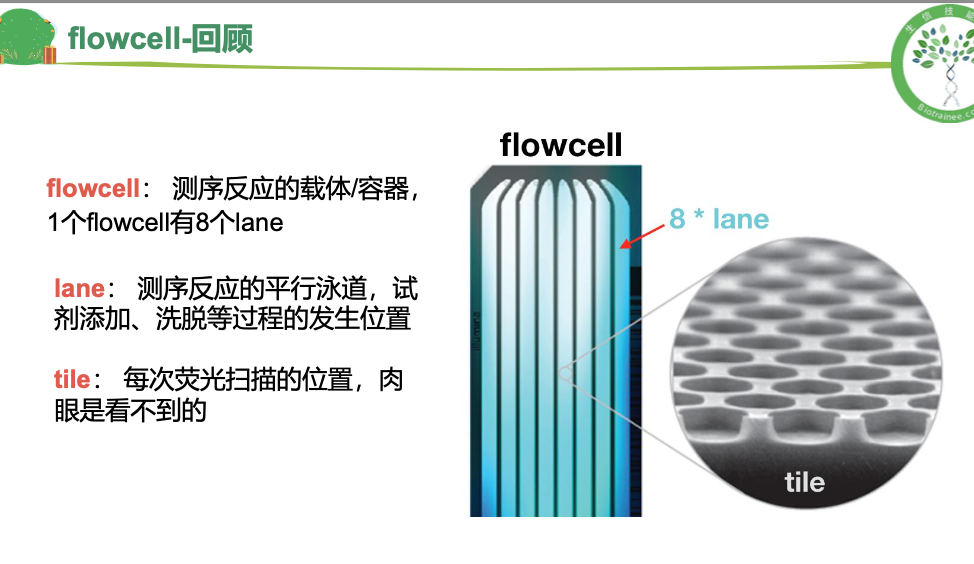<a name="K0DVo"></a>### 代码这里需要注意的是修改rawdata路径 以及```r# 激活conda环境conda activate rna# 连接数据到自己的文件夹cd $HOME/project/Human-16-Asthma-Trans/data/rawdataln -s /home/t_rna/data/airway/fastq_raw25000/*gz ./ 之前已经连接过这里就不用了# 使用FastQC软件对单个fastq文件进行质量评估,结果输出到qc/文件夹下nohup fastqc -t 6 -o ./ SRR*.fastq.gz >qc.log &# 使用MultiQc整合FastQC结果# 使用绝对路径运行multiqc=/home/t_rna/miniconda3/envs/rna/bin/multiqcfastqc=/home/t_rna/miniconda3/envs/rna/bin/fastqcfq_dir=/home/t_rna/project/Human-16-Asthma-Trans/data/rawdataoutdir=/home/t_rna/project/Human-16-Asthma-Trans/data/rawdata#这里的两个fq_dir 和 out_dir 等于的东西是自己rawdata路径$fastqc -t 6 -o $outdir ${fq_dir}/SRR*.fastq.gz >${fq_dir}/qc.log$multiqc $outdir/*.zip -o $outdir/ >${fq_dir}/multiqc.log
数据过滤

trim_galore过滤
基本概念


代码
这里cleandata和rawdata变量命名 是没有空格的
比如说课上联系 是这样的
fq_dir=/trainee/Apr2/project/Human-16-Asthma-Trans/data/rawdata
outdir=/trainee/Apr2/project/Human-16-Asthma-Trans/data/rawdat
# 激活小环境
conda activate rna
# 新建文件夹trim_galore
cd $HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
# 先生成一个变量,为样本ID
ls $HOME/project/Human-16-Asthma-Trans/data/rawdata/*_1.fastq.gz | awk -F'/' '{print $NF}' | cut -d'_' -f1 >ID
#这里要先看好自己的数据 最后保存到ID
# 多个样本 vim trim_galore.sh,以下为sh的内容
rawdata=$HOME/project/Human-16-Asthma-Trans/data/rawdata
cleandata=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
cat ID | while read id
do
trim_galore -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ${cleandata} ${rawdata}/${id}_1.fastq.gz ${rawdata}/${id}_2.fastq.gz
done
# 提交任务到后台
nohup sh trim_galore.sh >trim_galore.log & #把任务动态写在日志里帮助回忆
##这一步其实是把之前写的脚本运行并提交后台并写日志
# 使用MultiQc整合FastQC结果
multiqc *.zip
## ==============================================
## 补充技巧:使用掐头去尾获得样本ID
ls $rawdata/*_1.fastq.gz | while read id
do
name=${id##*/}
name=${name%_*}
echo "trim_galore -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ${cleandata} ${rawdata}/${name}_1.fastq.gz ${rawdata}/${name}_2.fastq.gz "
done
这里可以写个脚本(避免太多了忘记) 用vim写<br />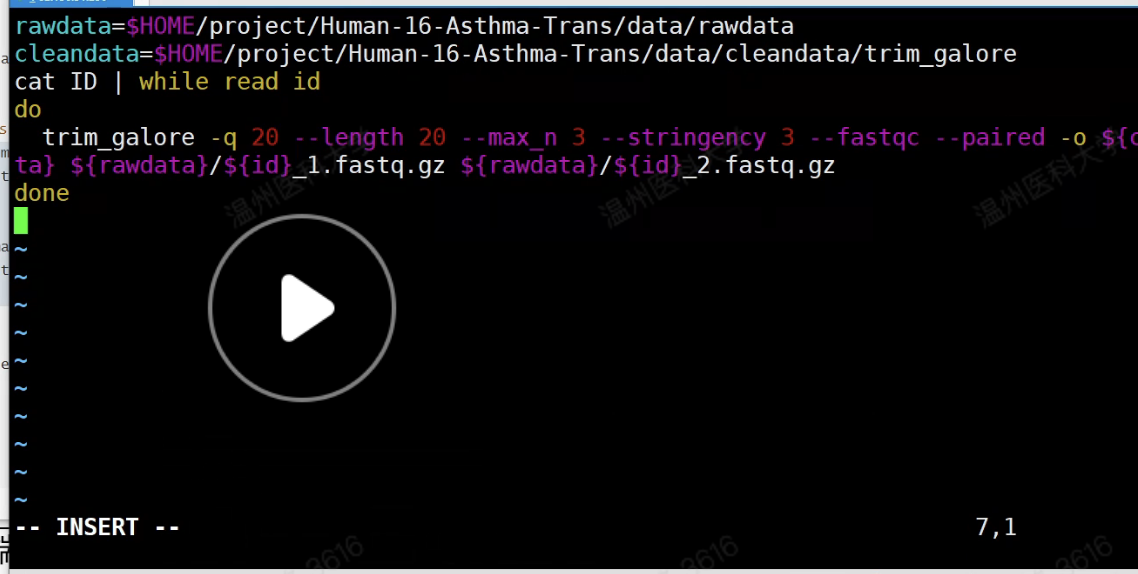esc-:-wq保存<br />可以使用cat来查看刚刚写的脚本<br />
这里值得注意的是,有的服务器是qsub,所以把命令echo到脚本里,然后用qsub运行<br />
<a name="rNlZ8"></a>
### fastp过滤
<a name="aKr0d"></a>
#### 基本概念
<a name="mv5Ow"></a>
#### 代码
注意这里也是cleandata和rawdata的修改成自己的,以及在数据很大的时候建议使用fastq这种方式
```r
cd $HOME/project/Human-16-Asthma-Trans/data/cleandata/fastp
# 定义文件夹:vim fastp.sh 以下是脚本内容
cleandata=$HOME/project/Human-16-Asthma-Trans/data/cleandata/fastp/
rawdata=$HOME/project/Human-16-Asthma-Trans/data/rawdata/
cat ../trim_galore/ID | while read id
do
fastp -l 36 -q 20 --compression=6 \
-i ${rawdata}/${id}_1.fastq.gz \
-I ${rawdata}/${id}_2.fastq.gz \
-o ${cleandata}/${id}_clean_1.fq.gz \
-O ${cleandata}/${id}_clean_2.fq.gz \
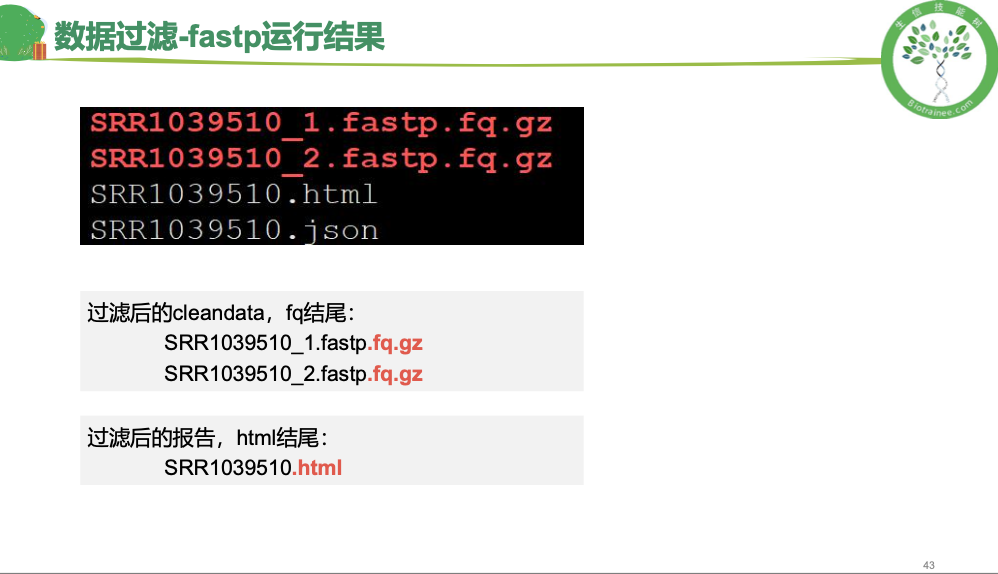
-R ${cleandata}/${id} \
-h ${cleandata}/${id}.fastp.html \
-j ${cleandata}/${id}.fastp.json
done
# 运行fastp脚本
nohup sh fastp.sh >fastp.log &
笔记记录
- 工作目录的管理十分重要


后续的分析也是在不同的目录下进行分析
值得注意的是 参考基因组数据库命名习惯 项目分析命名等等
- 创建好好文件夹后,到数据质控这一步,老师指导我们链接到服务器上的练习数据到rawdata文件夹里

这里切换位置——连接,可以ls -l一下 可以看到文件的来源
可以看到文件的来源
- 输入history | less可以显示出之前输出过的命令
- 很多脚本是写成sh脚本的
- 查看rawdata的ID特点
 其实就是获取SRR的这串数字不一样,也就是获取这段变量awk -F’/‘ ‘{print $NF}’ | cut -d’_’ -f1 >ID
其实就是获取SRR的这串数字不一样,也就是获取这段变量awk -F’/‘ ‘{print $NF}’ | cut -d’_’ -f1 >ID

 (这里重温awk)$NF 输出最后一行
(这里重温awk)$NF 输出最后一行
- fastp是写脚本 运行完了可以看一眼

- 最后输出是html网页文件,名字带有reporter 或cleandata 在服务器上,需要下载到自己的文件夹里再打开
以上代码和pdf文件内容均来自生信技能树课程

若有收获,就点个赞吧
0 人点赞
