最近在尝试做个高分子复合材料的建模,一开始用的MS被气得不轻。动不动就程序崩溃,体系一大就完全跑不动,几十分钟跑1%。最后只好换个方法用了EMC,20MB的大小在建模这块简直吊打MS。
1.EMC下载安装
EMC文件下载地址:
下载完解压后,在终端输入以下命令安装调用EMC的python库:
pip install emc-pypi
在python中执行以下代码,没有报错则说明改库安装成功。
import pyemc
由于EMC主体是用perl写的,所以电脑还需要支持perl环境,在终端输入以下命令查看是否支持perl:
perl -v
输出如下则表明已安装perl:
如果提示找不到perl,自行上官网下载安装就行了。
2.esh文件编写
下载的文件中自带了一些esh的例子,照着修改可以了。我用于生成PET/PE复合材料的esh如下:
ITEM OPTIONSreplace true #覆盖存在的脚本mass true #在data中添加质量ntotal 7200 #体系原子总数density 0.3 #体系密度field pcff #设置力场(EMC支持COMPASS, PCFF, CHARMM, OPLS, TraPPE or coarse-grained force fields)build_dir . #我也不知道有啥用ITEM ENDITEM SHORTHAND #提供了一种快速构建化学文件的方法PET O=C(C1=CC=C(C(OCCOC(C2=CC=C(C(OCCOC(C3=CC=C(C(OCCOC(C4=CC=C(C(OCCOC(C5=CC=C(C(OCCOC(C6=CC=C(C(OCCOC(C7=CC=C(C(OCCOC(C8=CC=C(C(OCCOC(C9=CC=C(C(OCCOC(C%10=CC=C(C(C)=O)C=C%10)=O)=O)C=C9)=O)=O)C=C8)=O)=O)C=C7)=O)=O)C=C6)=O)=O)C=C5)=O)=O)C=C4)=O)=O)C=C3)=O)=O)C=C2)=O)=O)C=C1)OCCOC,10 #名称,SMILE式,链数PE CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC,20 #名称,SMILE式,链数ITEM ENDITEM COMMENTS #设置体系组分,含量ITEM GROUPS #组分PET O=C(C1=CC=C(C(OCCOC(C2=CC=C(C(OCCOC(C3=CC=C(C(OCCOC(C4=CC=C(C(OCCOC(C5=CC=C(C(OCCOC(C6=CC=C(C(OCCOC(C7=CC=C(C(OCCOC(C8=CC=C(C(OCCOC(C9=CC=C(C(OCCOC(C%10=CC=C(C(C)=O)C=C%10)=O)=O)C=C9)=O)=O)C=C8)=O)=O)C=C7)=O)=O)C=C6)=O)=O)C=C5)=O)=O)C=C4)=O)=O)C=C3)=O)=O)C=C2)=O)=O)C=C1)OCCOC #名称,smile式PE CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC #名称,smile式ITEM ENDITEM CLUSTERS #含量PET PET,5 #名称,名称,链数PE PE,20 #名称,名称,链数ITEM END #含量
将上面这段代码保存为1.esh。
smile式在许多化学资料库上可以查找,但是还是会有很多缺少的。建议还是自己用chemdraw绘制并导出smile式。
3.执行EMC生成data文件
在python中运行以下程序
import pyemcpyemc.setup('xxx.esh')pyemc.build('build.emc')
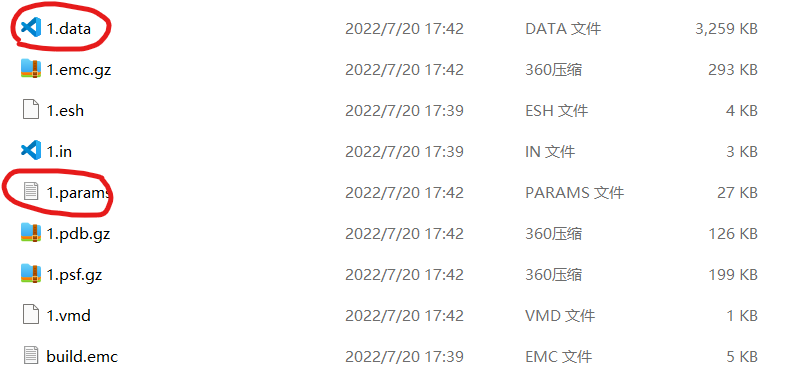
4.文件详情
params文件中主要包含模型的势函数,质量等参数:
......# Massesmass 1 12.01115 # cmass 2 12.01115 # c_0mass 3 12.01115 # c_1mass 4 12.01115 # cpmass 5 1.00797 # hcmass 6 15.99940 # o_1mass 7 15.99940 # o_2mass 8 15.99940 # oc# Potentialspair_style lj/class2/coul/long ${cutoff} ${charge_cutoff} # 9.5bond_style class2angle_style class2dihedral_style class2improper_style class2pair_modify mix sixthpower tail yesspecial_bonds lj/coul 0 0 1# Pair Coeffspair_coeff 1 1 0.05400 4.01000 # c,cpair_coeff 2 2 0.12000 3.30800 # c_0,c_0pair_coeff 3 3 0.12000 3.81000 # c_1,c_1pair_coeff 4 4 0.06400 4.01000 # cp,cppair_coeff 5 5 0.02000 2.99500 # hc,hcpair_coeff 6 6 0.26700 3.30000 # o_1,o_1pair_coeff 7 7 0.24000 3.42000 # o_2,o_2pair_coeff 8 8 0.24000 3.53500 # oc,oc......
data文件详情如下:
LAMMPS output created by EMC v9.4.4, build Aug 20 2021 07:52:527140 atoms7185 bonds13760 angles19981 dihedrals8520 impropers8 atom types12 bond types20 angle types26 dihedral types15 improper types0 60.21913826 xlo xhi0 60.21913826 ylo yhi0 60.21913826 zlo zhiMasses1 12.01115 # c2 12.01115 # c_03 12.01115 # c_14 12.01115 # cp5 1.00797 # hc6 15.99940 # o_17 15.99940 # o_28 15.99940 # ocAtoms1 1 6 -0.5310 42.9929423661 30.7556164498 8.4423851246 # o_12 1 3 0.7200 41.8686922004 31.1522224122 8.7061678569 # c_13 1 4 -0.0180 40.5796544532 30.6967084270 8.0664396047 # cp4 1 4 -0.1268 40.6052652233 29.6647885167 7.1066772045 # cp5 1 5 0.1268 41.5743523233 29.1512023110 6.8297214122 # hc......
将data导入ovito中:

若有收获,就点个赞吧
0 人点赞
