- grep
- 正则表达式
- 查找pattern
- cat example.fa | grep -n ‘^ATATAA’
- cat example.fa | grep -n ‘^ATATAA’
- cat example.fa | grep -n ‘.ATATAA$’ 模糊查找
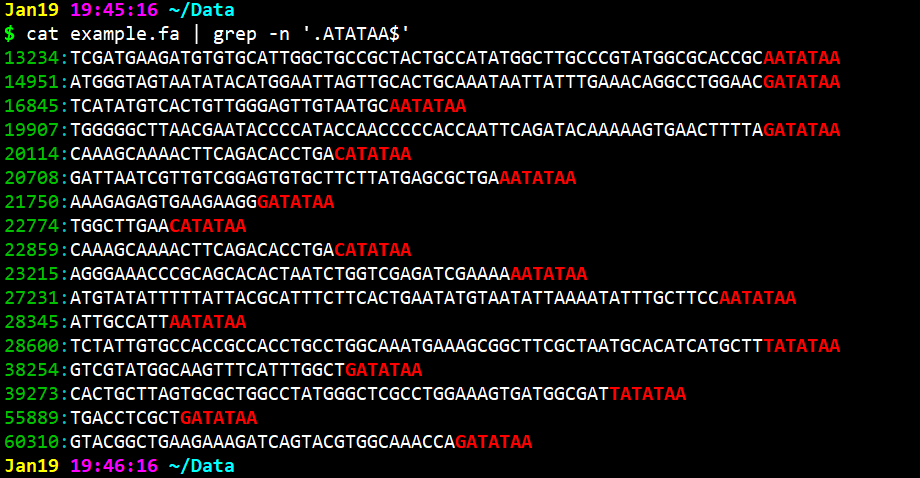
- cat readme.txt | grep -E ‘f?ee’ -E的意思是正则表达式
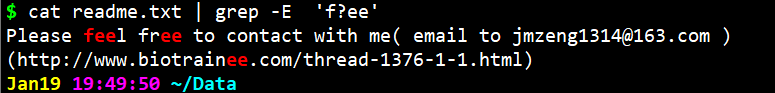
- $ cat readme.txt | grep ‘f\?ee’
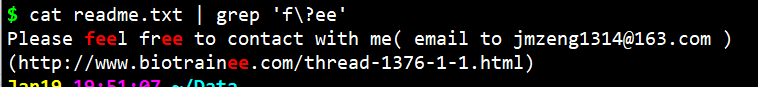
- cat readme.txt | grep ‘re+‘
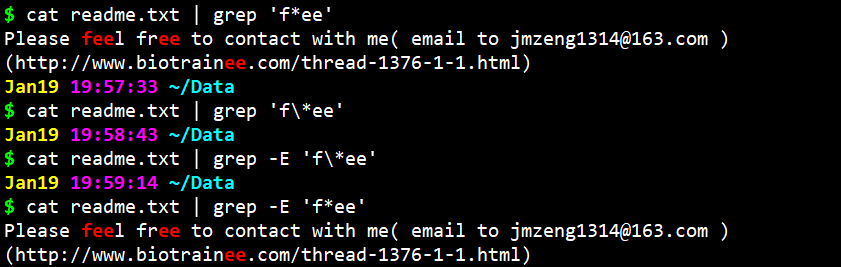
- cat readme.txt | grep ‘f*ee’
- cat readme.txt | grep [bB]
- 练习题
- 过滤:cat example.fq | grep ‘^@’ | grep -v ‘^@ERR’
- $ cat example.gtf | grep -w -E ‘CDS|UTR|exon’ | less -SN
- sed 流编辑器,一般用来对文本进行增删改查
- 用法
- cat readme.txt | sed ‘1 a Hi’
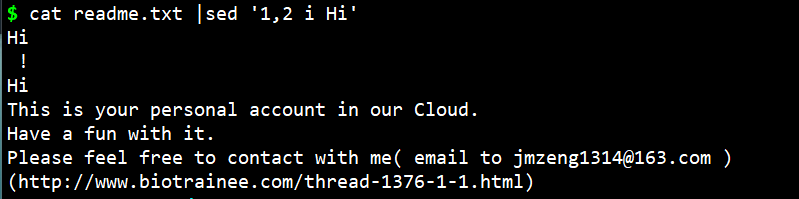
- cat readme.txt |sed ‘1,2 i Hi’
- $ less -SN file3 | sed ‘1d’ | less -SN
- cat readme.txt |sed -e ‘2i Hi’ -e ‘2a Hi’
- $ cat readme.txt |sed -e ‘2,4c *‘
- 如何插入多行:cat readme.txt |sed -e ‘2,4c *\n*\n*‘
- cat readme.txt |sed -e ‘s/is/IS/‘
- $ cat readme.txt |sed ‘s+is+IS+’ 用别的符号代替/也可以
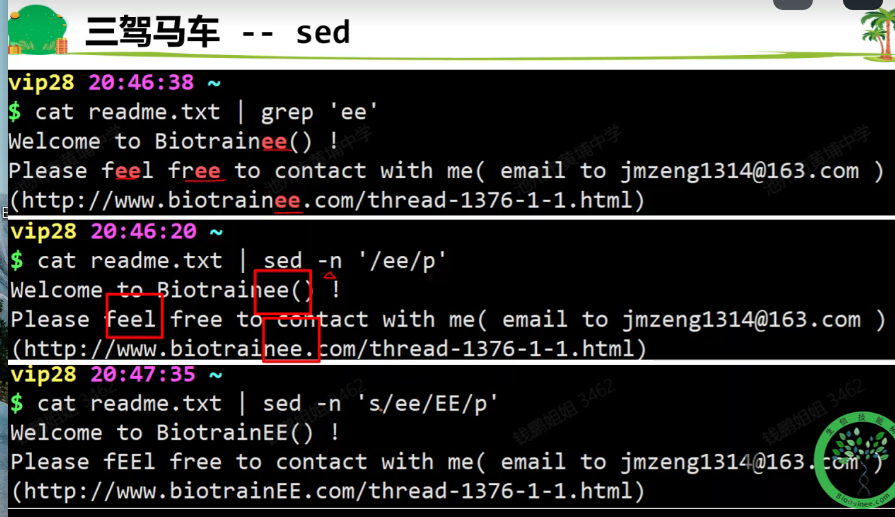
- cat readme.txt | sed ‘1~3s/ee/EE/‘ 第一行开始每隔三行改一行
- 查找
- 作业
- awk :编程语言,可对文本和数据进行处理
grep
查找pattern 并将其以行号的形式输出
grep -r 'ATAAT' . -n

精确查找gene的pattern,精确查找指的是,完全包含某个单词
``
less Data/example.gtf | grep -w ‘gene’ 
精确查找gene的pattern并按行输出
less example.gtf | grep -w ‘gene’ | less -SN 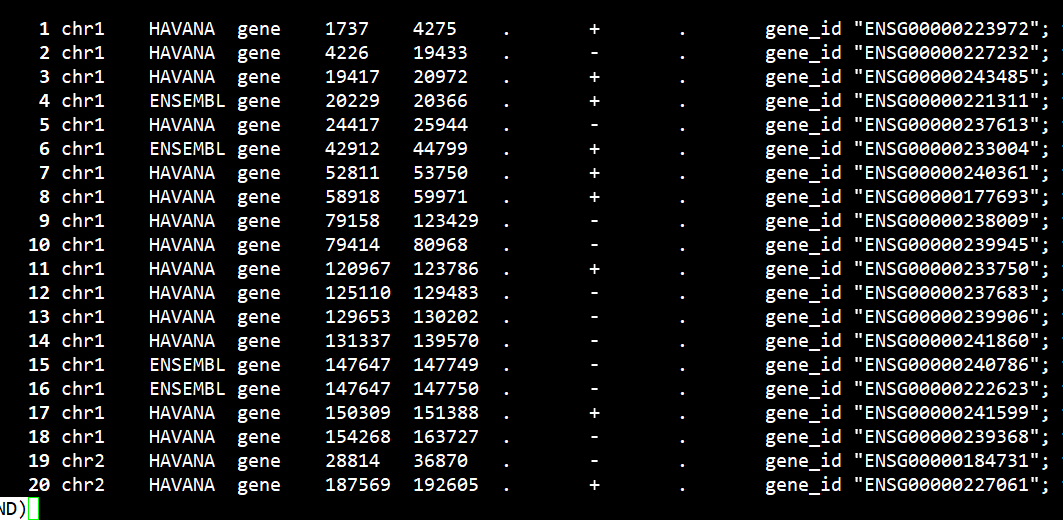
$ less Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | grep -w ‘gene’ | less -SN 并没有精确拿出第三列是gene 的一行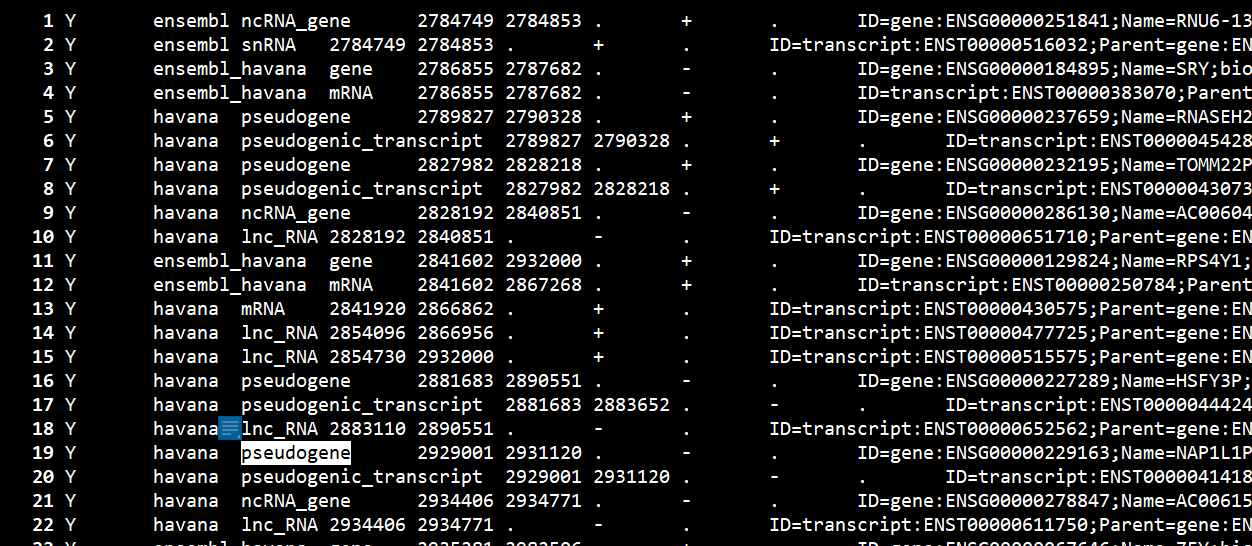
过滤掉所有含”gene” 的行
$ less example.gtf | grep -w -v ‘gene’ 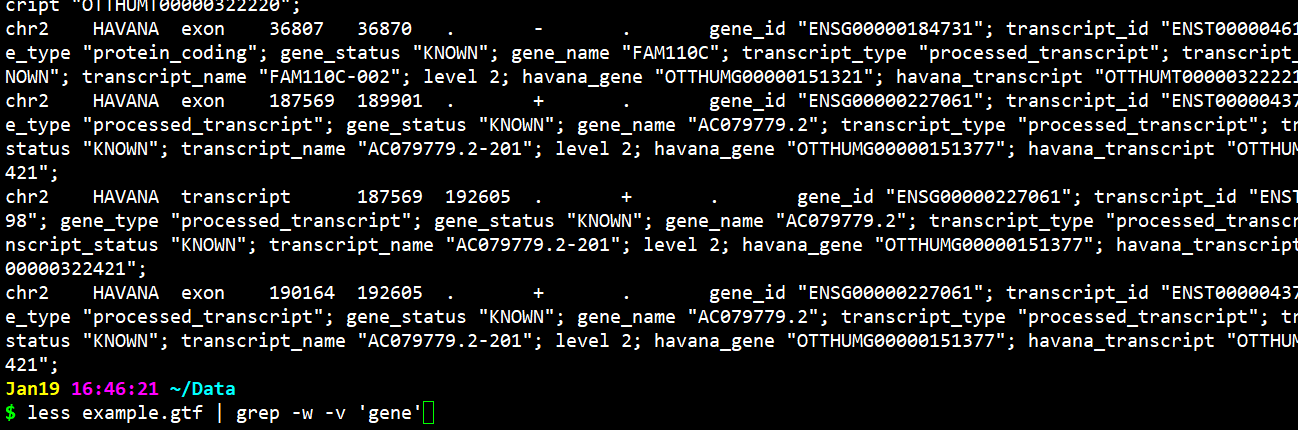
统计没有“gene” 的行数
$ less example.gtf | grep -w -v -c ‘gene’
统计所有的行数,并验算
$ wc -l example.gtf
237 example.gtf
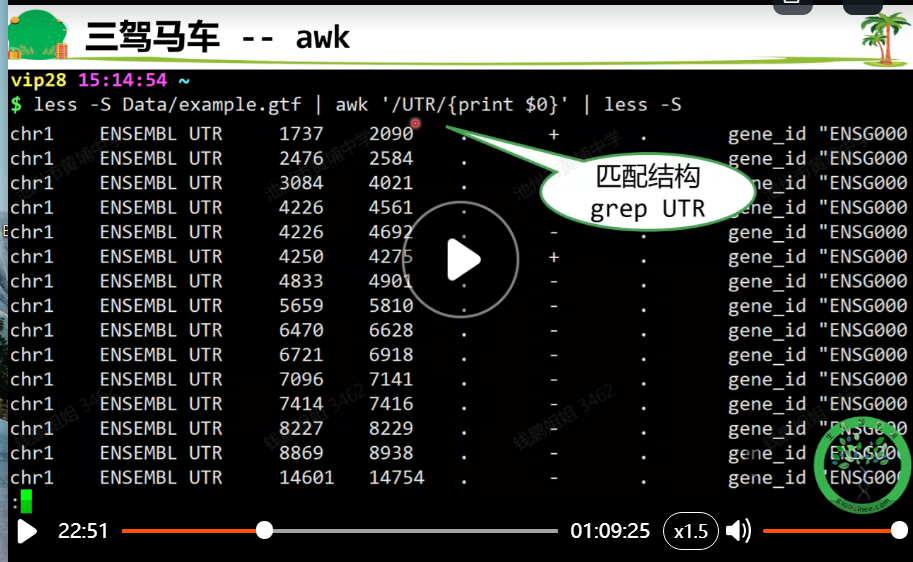
查找含有’gene’或者’UTR’的pattern
less example.gtf | grep -w -e 'gene' -e 'UTR'
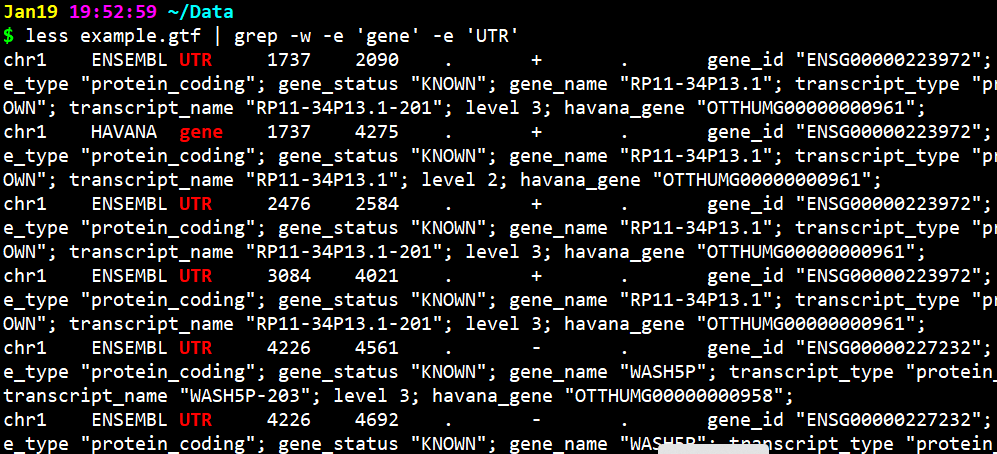
如何进行多个pattern的查找(>2个)
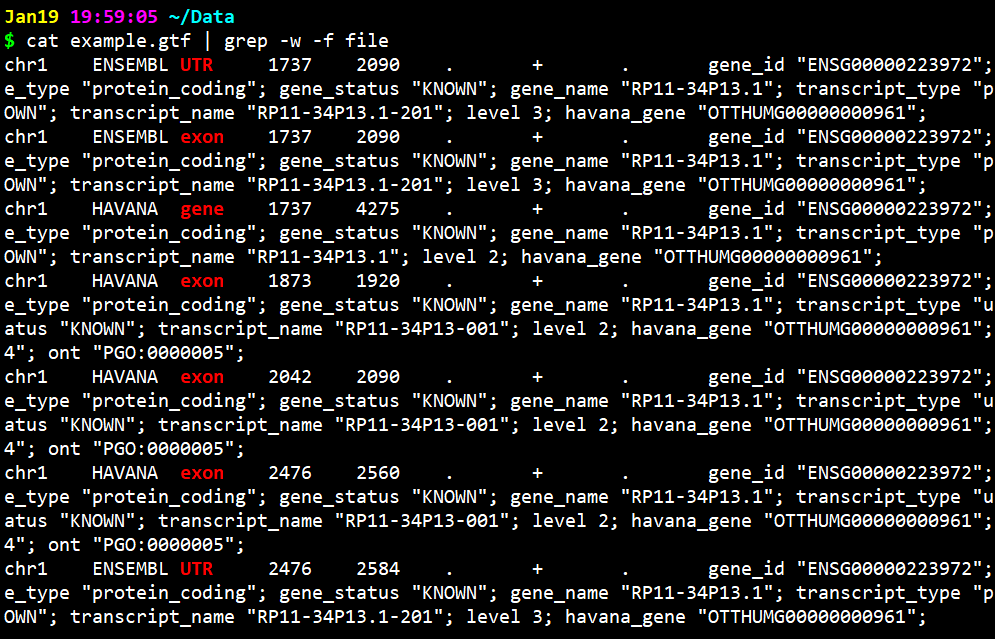
cat > fileUTRgeneexon^Ccat example.gtf | grep -w -f filesecond metholdless example.gtf | grep -w -f file | less -S
正则表达式

查找pattern
cat example.fa | grep -n ‘^ATATAA’
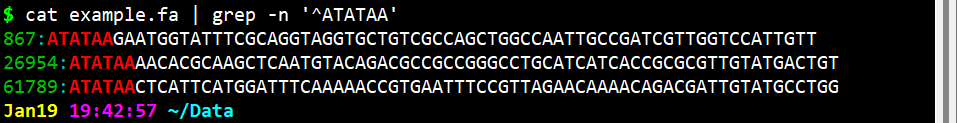
cat example.fa | grep -n ‘^ATATAA’
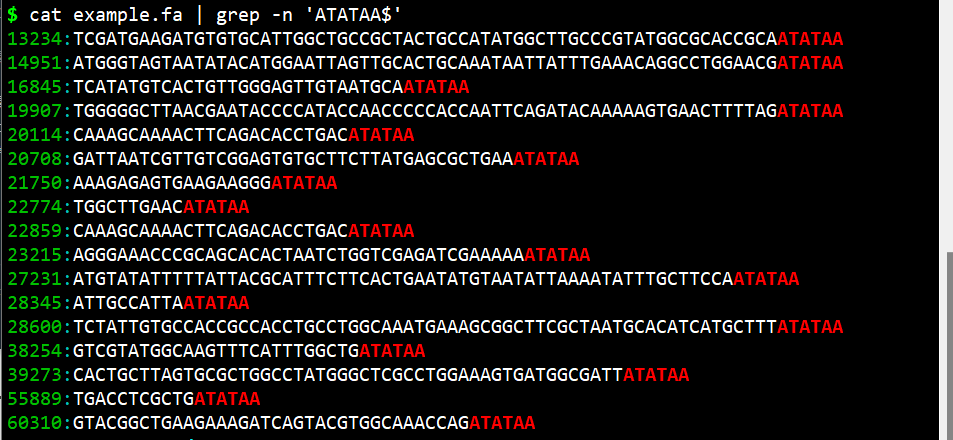
cat example.fa | grep -n ‘.ATATAA$’ 模糊查找
cat readme.txt | grep -E ‘f?ee’ -E的意思是正则表达式
$ cat readme.txt | grep ‘f\?ee’
cat readme.txt | grep ‘re+‘

cat readme.txt | grep ‘f*ee’

cat readme.txt | grep [bB]

练习题

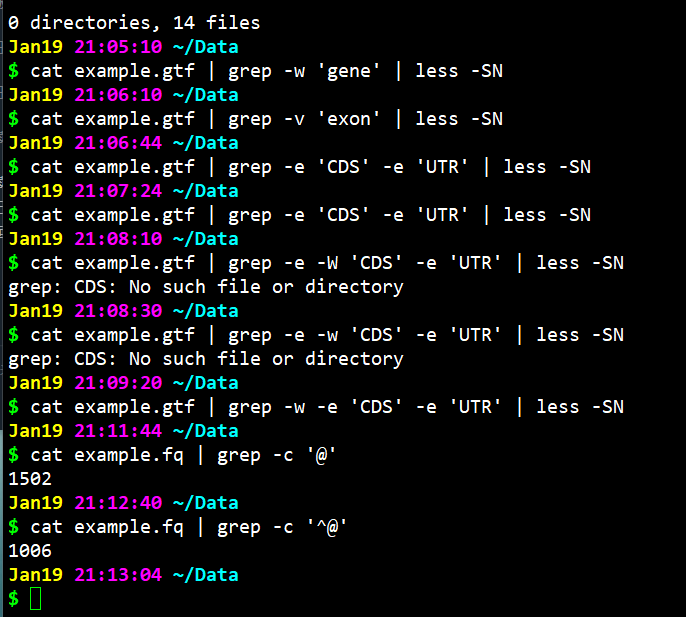
过滤:cat example.fq | grep ‘^@’ | grep -v ‘^@ERR’
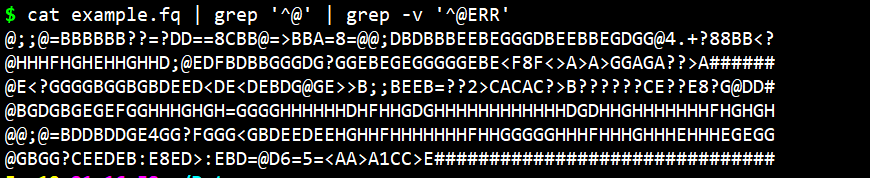
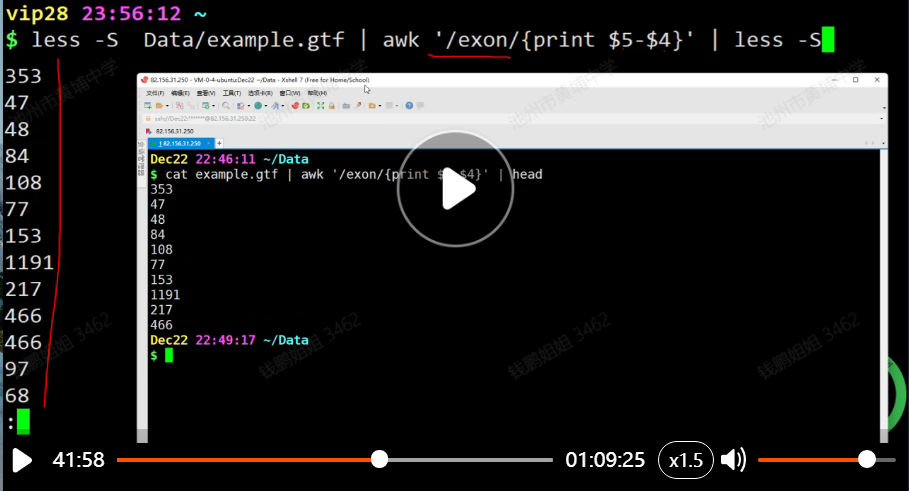
$ cat example.gtf | grep -w -E ‘CDS|UTR|exon’ | less -SN

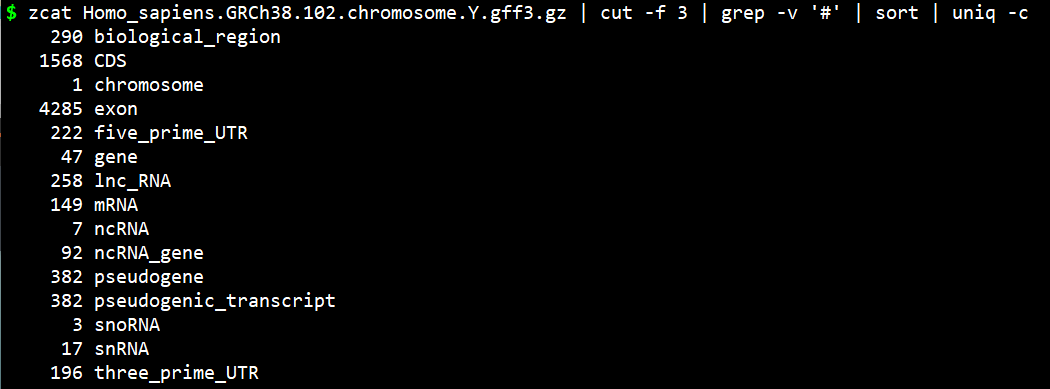
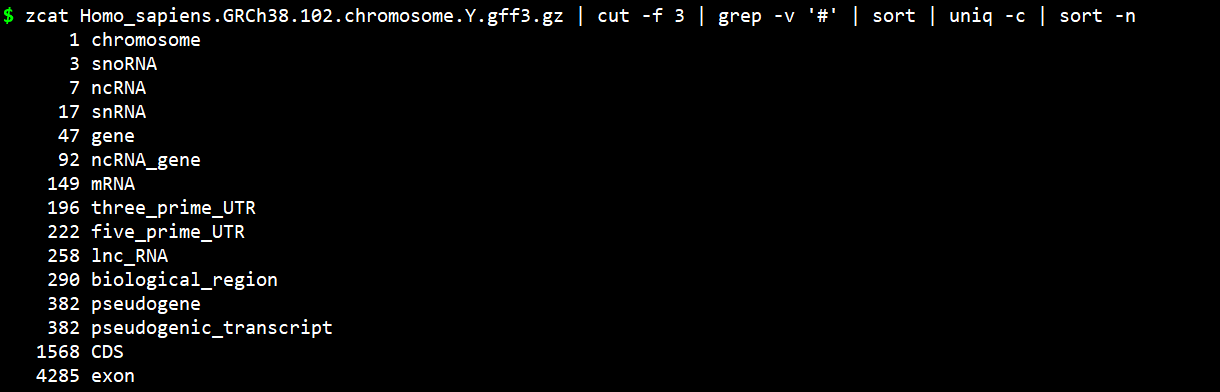
$ zcat Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | cut -f 3 | grep -v '#' | less$ zcat Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | cut -f 3 | grep -v '#' | less | grep -w 'gene' -c47
sed 流编辑器,一般用来对文本进行增删改查

用法
[!]:是取反的意思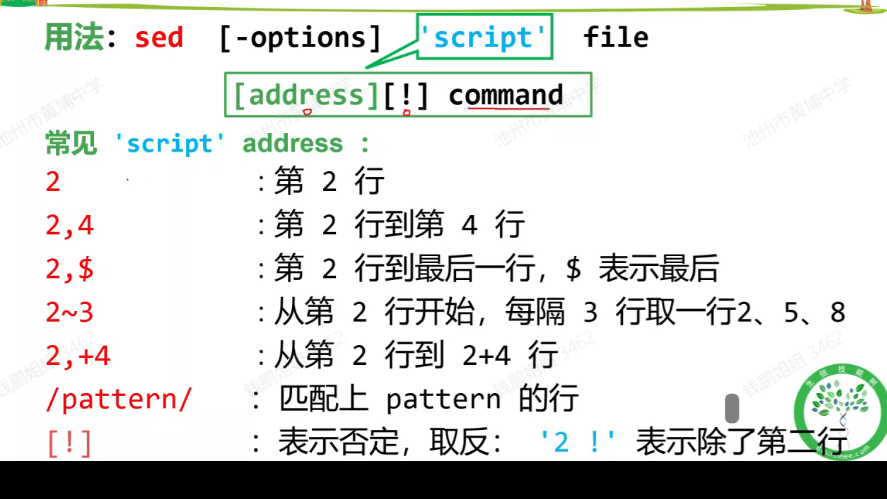
vim 中加%是对某一行进行处理
sed默认对所有行处理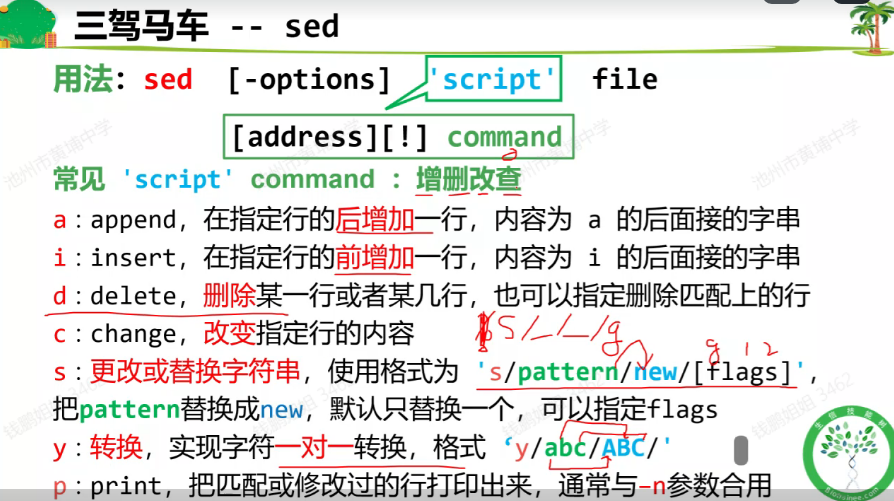
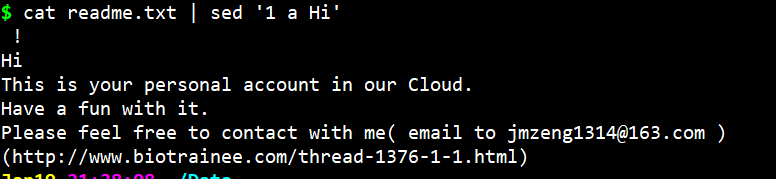
cat readme.txt | sed ‘1 a Hi’
cat readme.txt |sed ‘1,2 i Hi’
$ less -SN file3 | sed ‘1d’ | less -SN
删除第一行
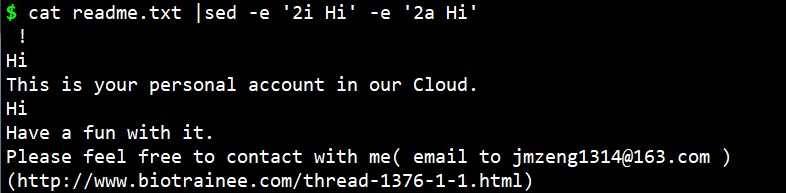
cat readme.txt |sed -e ‘2i Hi’ -e ‘2a Hi’
$ cat readme.txt |sed -e ‘2,4c *‘
更改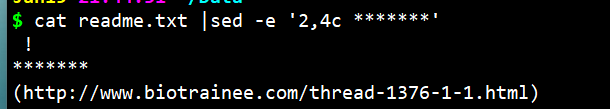
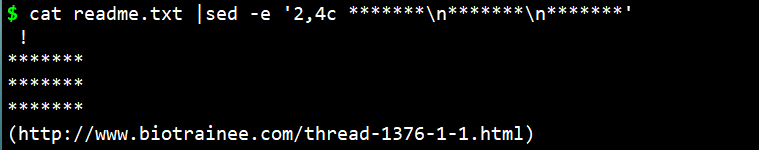
如何插入多行:cat readme.txt |sed -e ‘2,4c *\n*\n*‘
cat readme.txt |sed -e ‘s/is/IS/‘
$ cat readme.txt |sed ‘s+is+IS+’ 用别的符号代替/也可以
cat readme.txt | sed ‘1~3s/ee/EE/‘ 第一行开始每隔三行改一行

查找
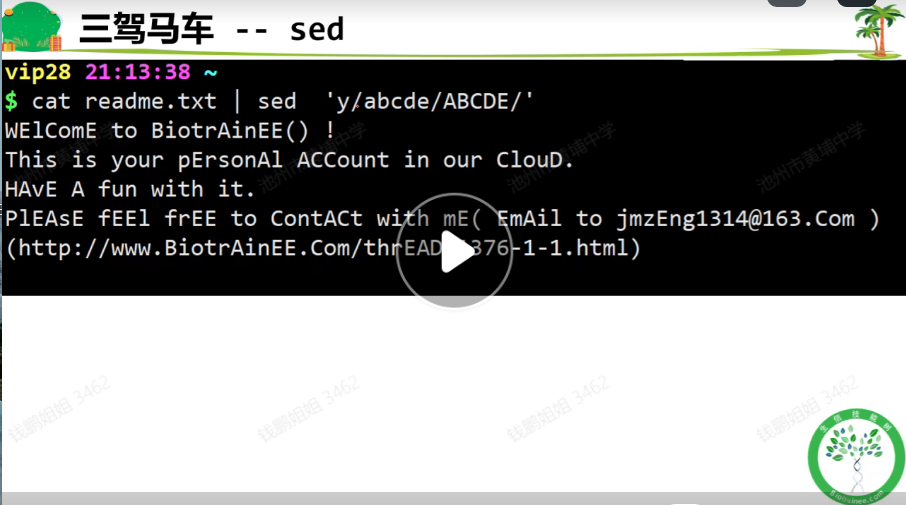

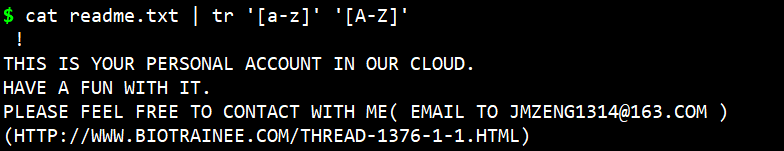
大小写替换
作业

$ head example.gtf | sed 's/HAVANA/ENSEMBL/' | less -SNhead example.fa | sed -n '2p' | sed 'y/ATCG/TAGC/' | rev (方向互补)
awk :编程语言,可对文本和数据进行处理

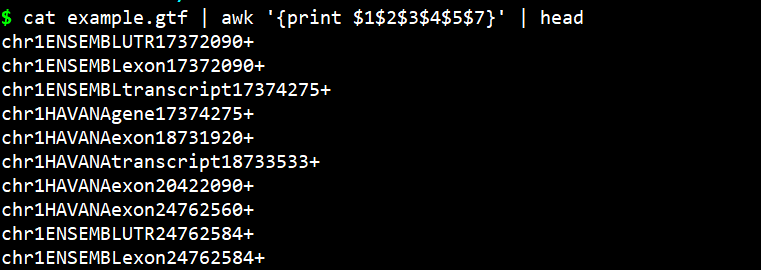
awk 只能拿出一块,但是cut 只能识别 \t (Table键) 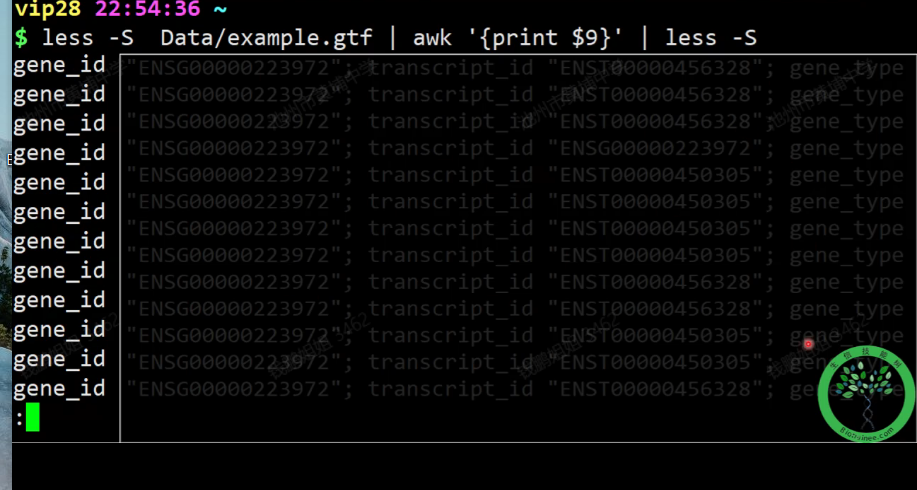
观察:一个是空格,一个是制表符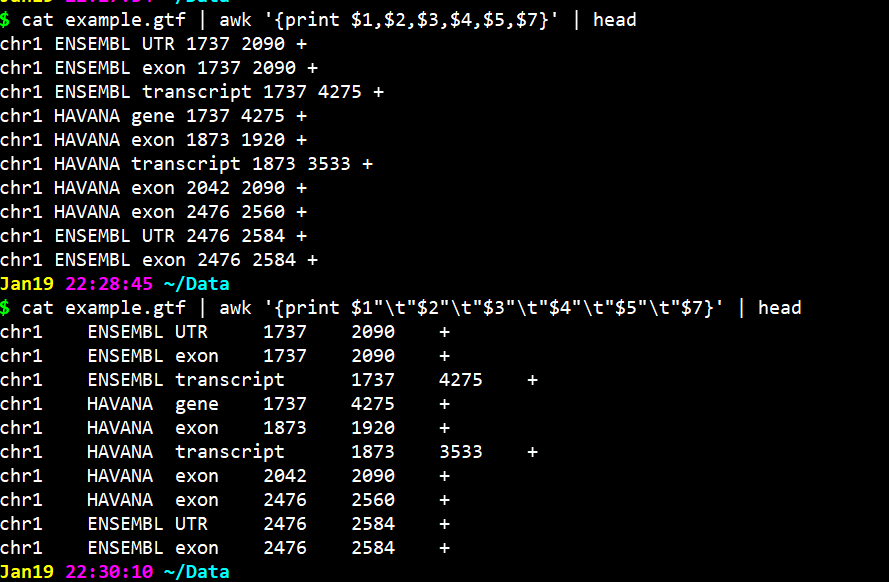

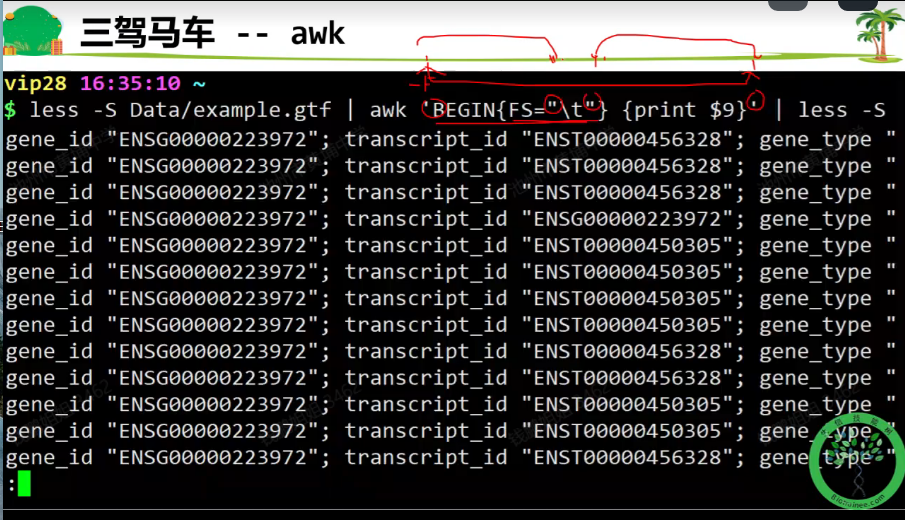
内置变量

设置分隔符
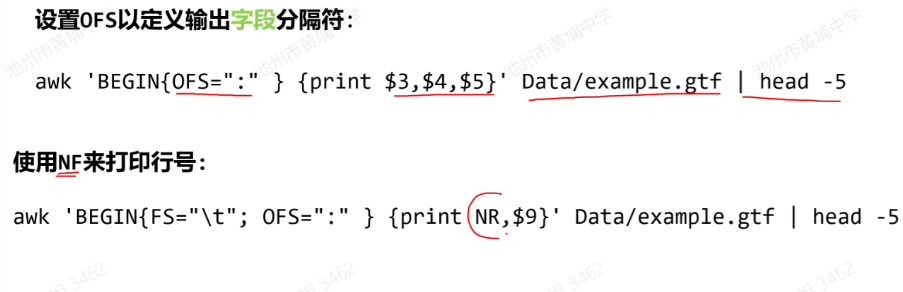
FS定义字段分隔符
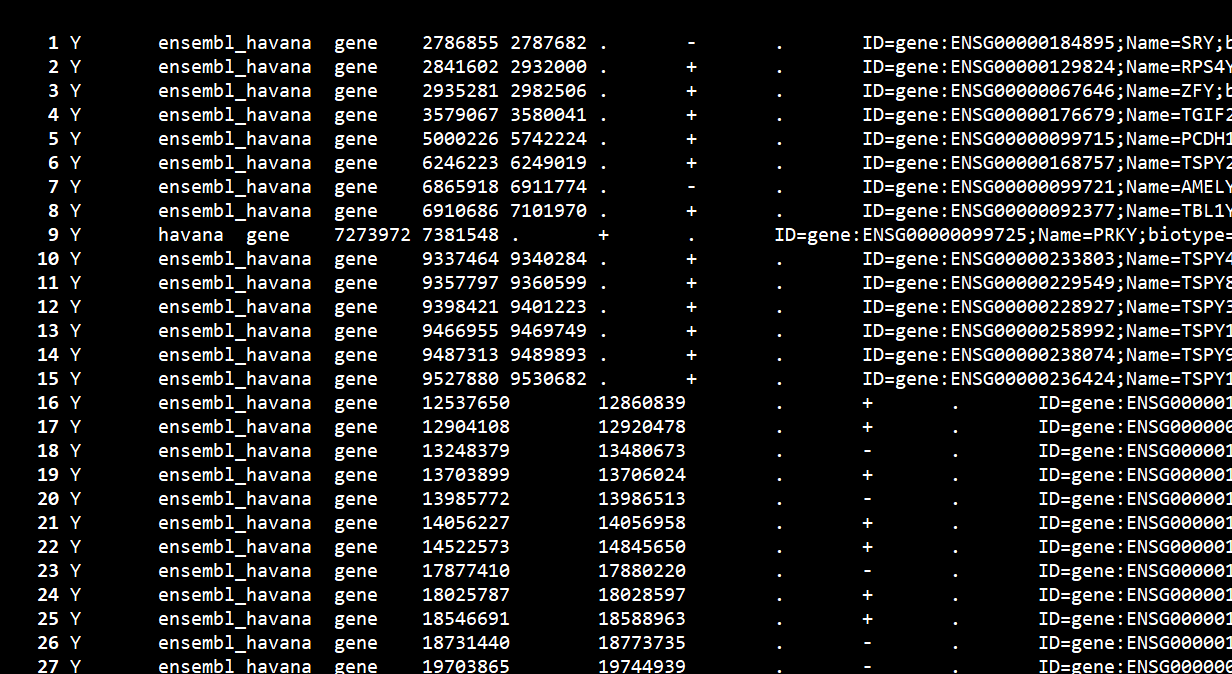
从测试文件中拿出Y染色体匹配的gene(完全匹配) 的行
zcat Homo_sapiens.GRCh38.102.chromosome.Y.gff3.gz | awk '{if($3=="gene")print $0}' | less -SN
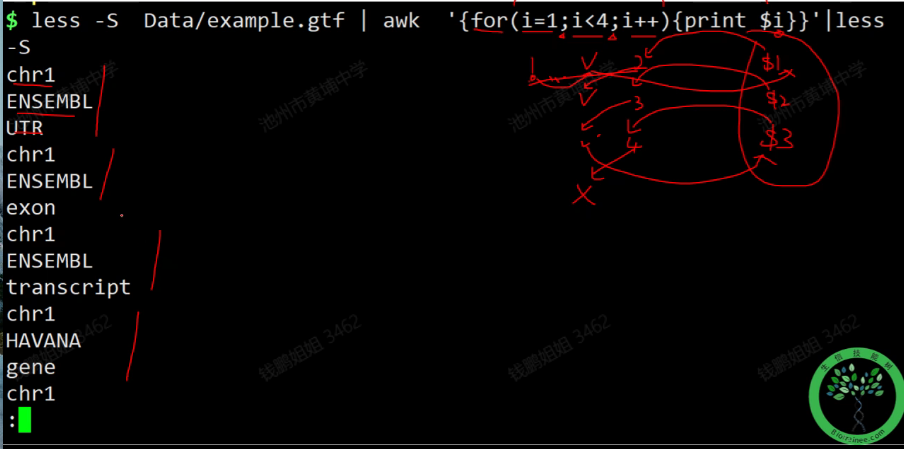
FOR 循环
awk的数学运算

作业

若有收获,就点个赞吧
0 人点赞
