一,数据获得:
TCGA:https://xenabrowser.net/datapages/?cohort=GDC%20TCGA%20Colon%20Cancer%20(COAD)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) 来自UCSC xena数据库的 TCGA数据,表达值为fpkm,共有512个样本,已经过log2(fpkm+1)标准化,对应的临床数据也从改数据库中下载获得。对得到的临床数据进行筛选(去除掉没有生存信息和生存时间小于30天的样本后),最终剩余419个样本进行后续研究。
GSE17538:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi 来自GEO的芯片数据,使用R包 affy的RMA方法进行预处理后(内部log2() 进行标准化)。平台为GPL570(HG-U133_Plus_2),该平台为探针芯片平台,需要对探针进行重注释才能找到相应的lncRNA,因此根据我们已有的probe-lncRNA的GPL570 重注释文件从而提取lncRNA表达谱。对os_time进行筛选,过滤掉无生存信息和生存时间小于30天的样本后,最终剩下219**个样本进行后续研究。
二,mRNA注释文件:https://www.genenames.org/ 从HGNC数据库中下载得到,目的是为了从COAD数据中筛选出mRNA表达谱,并且把entrez gene ID 转换成 gene symbol,最终得到 419个样本的 37393 个mRNA的表达谱文件进行后续研究。
lncRNA注释文件:https://www.genenames.org/ 从HGNC数据库中下载得到,目的是为了从COAD数据中筛选出lncRNA表达谱,并且过滤掉表达值为0在总体样本中占20%以上的lncRNA,最终得到 419个样本的 3959 个lncRNA的表达谱文件进行后续研究。
GEO芯片数据重注释规则:当一个探针只与一个lncRNA对应时,该lncRNA的表达水平为相对应唯一探针的表达水平;当多个探针与一个lncRNA对应时,该lncRNA的表达水平为相应多个探针表达水平的均值;当一个探针对应多个lncRNA的时候,该情况删除。通过重注释步骤能重注释出5919个lncRNA表达谱文集,并且与TCGA的lncRNA表达谱(20%过滤后)取交集后,筛选待研究的lncRNA表达谱数据(n=2235)
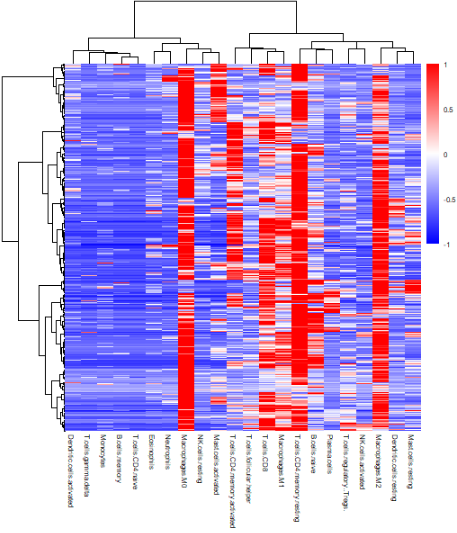
三,免疫浸润评估:https://cibersort.stanford.edu/ 使用来自斯坦福大学开发的计算工具 CIBERSORT 进行免疫细胞浸润的评估,评估了22种免疫细胞在COAD的浸润分值,permutation = 500(计算的时候排列500次)。免疫细胞具体详情列在表格中。热图表示了免疫细胞的浸润程度(蓝色代表低浸润,红色代表高浸润),发现了不同的免疫细胞类型在样本中呈现不同的浸润水平,有研究表明,免疫细胞的浸润程度是影响患者预后水平的关键因素,因此进一步探讨了与免疫细胞浸润相关的lncRNA。
为了更直观的展示数据在热图上的分布,我们对数据进行了scale()方法标准化,根据各自样本的均值和标准差计算出标准值,再进行热图的绘制。热图的绘制均使用pheatmap()方法,clustering_method选用的是ward.D方法。
| B cells naive | B cells memory | Plasma cells | T cells CD8 | T cells CD4 naive | T cells CD4 memory resting | T cells CD4 memory activated | T cells follicular helper | T cells regulatory (Tregs) | T cells gamma delta | NK cells resting | NK cells activated | Monocytes | Macrophages M0 | Macrophages M1 | Macrophages M2 | Dendritic cells resting | Dendritic cells activated | Mast cells resting | Mast cells activated | Eosinophils | Neutrophils |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
根据22种免疫细胞的浸润水平和lncRNA表达谱,使用R中的cor.test()方法计算每个免疫细胞浸润水平和每个lncRNA表达水平之间的相关性,并且根据p-value和peason相关系数进行筛选(p<0.05, |pearson cor|>0.3),最终筛选得到258**个**与免疫细胞浸润强相关的lncRNA。下热图表示了免疫细胞与lncRNA的相关性(色柱表示 |pearson| 相关系数)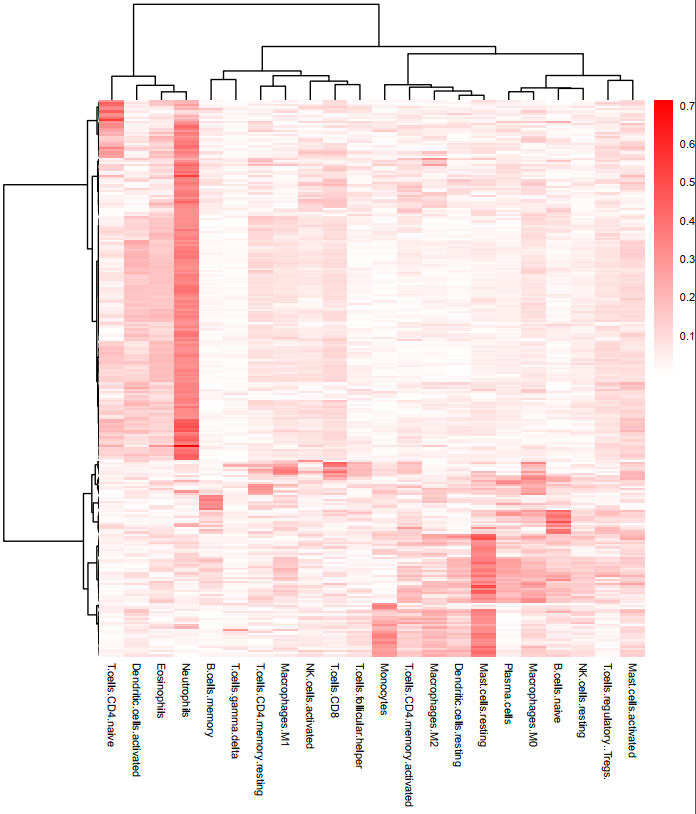
四,为了寻找预后相关的lncRNA,我们使用了全体TCGA COAD (n=419)的集合惊醒下列分析:
与生存相关的lncRNA筛选:使用R包(survival),使用coxph方法对258个lncRNA与全面生存时间(os time)和全面生存状态(os event)之间进行单因素风险回归,并选择p<0.05的lncRNA作为与COAD预后显著相关的lncRNA,得到**29个**lncRNA作为多cox筛选的候选基因。使用R包forestplot()对单cox步骤筛选出的lncRNA绘制森林图,展示每个lncRNA的HRs,CI95%,p-value。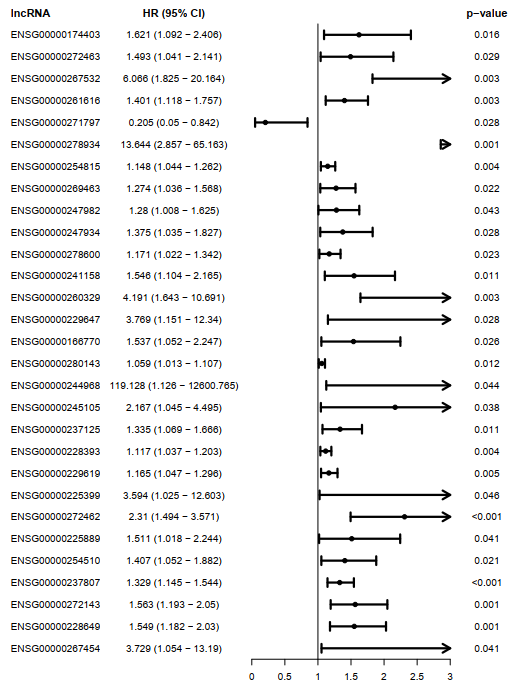
使用29个单cox预后相关的lncRNA结合临床因素(gender, age, stage)进行多cox回归,能矫正临床信息的影响,最终筛选得到4个矫正临床信息影响的预后相关的lncRNA。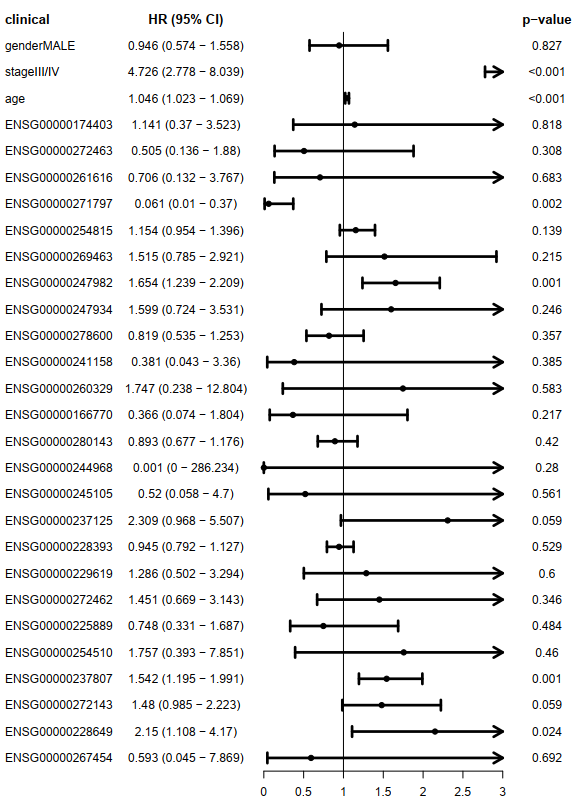
根据这4个lncRNA和对应的风险系数构建多cox模型,并根据各自的风险系数构建多cox风险回归模型。模型如下:
exp(ENSG00000271797)(-2.334119617) + exp(ENSG00000247982)0.289309684 + exp(ENSG00000237807)0.283336978 + exp(ENSG00000228649)0.579001809
使用R包forestplot对多cox步骤筛选出的lncRNA绘制森林图,展示每个lncRNA的HRs,CI95%,p-value。
ENSG00000271797属于保护因素(HR < 1);ENSG00000247982,ENSG00000237807,ENSG00000228649则属于危险因素(HR > 1)。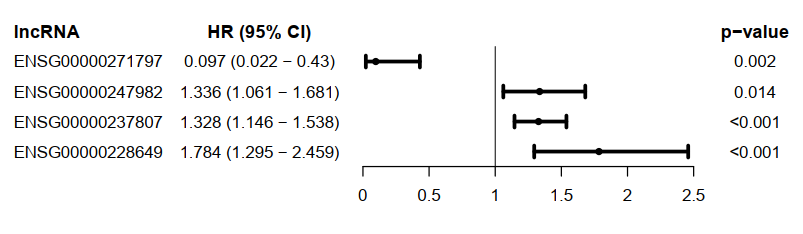
使用R包forestplot对多cox步骤筛选出的lncRNA结合其余临床信息绘制多cox森林图,展示每个lncRNA的HRs,CI95%,p-value。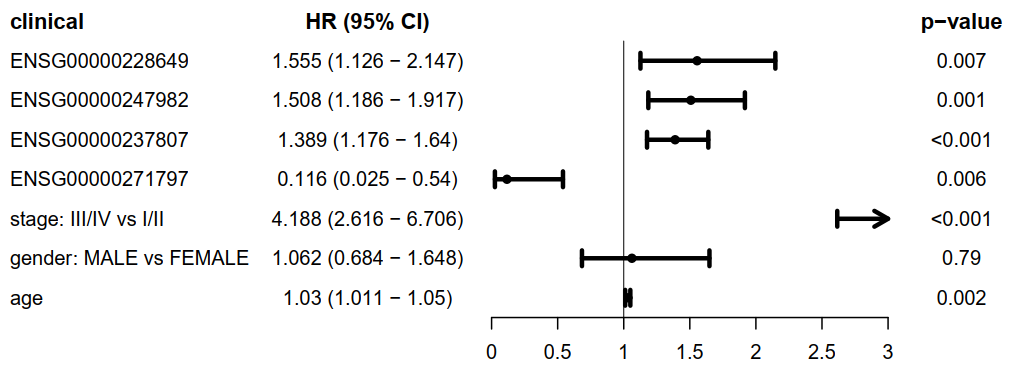
依据上述模型可以计算每个样本的风险得分,并根据训练集的中位值作为cutoff(-0.1805057**)**划分高低风险组,风险得分大于cutoff的定义为高风险组,风险得分小于cutoff的定义为低风险组,使用R包survminer 的ggsurvplot绘制KM曲线,log-rank的p<0.001。低风险组患者的预后显著优于高风险组。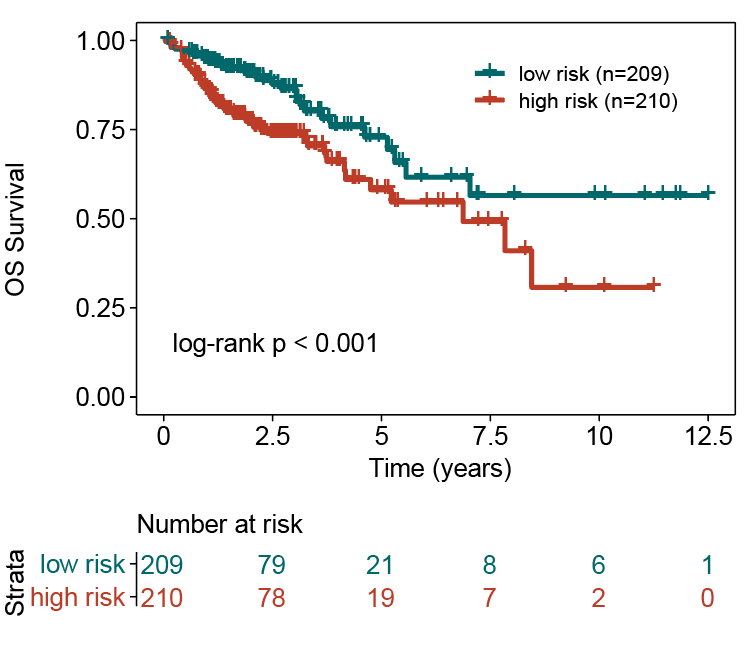
low risk:三年生存率:0.865 五年生存率:0.729
high risk: 三年生存率:0.739 五年生存率:0.581
为了体现风险模型如何预测数据集中的受试者的存活时间。特别是,假设我们有几天的生存时间,我们想看看标记如何预测三年/五年的存活(predict.time=3 years or 5 years)。该功能返回感兴趣的时间点的标记值、TP(真阳性)、FP(假阳性)、对应于感兴趣时间点(predict.time)和AUC(ROC)曲线下面积的Kaplan-Meier生存估计。使用R语言survivalROC进行上述操作方法。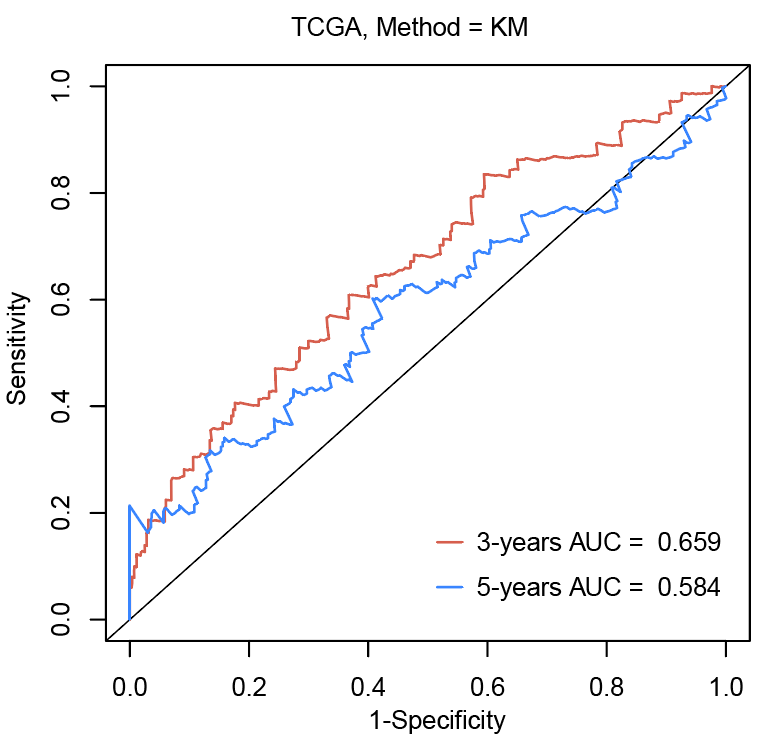
根据风险得分在训练集进行从低到高的排序,绘制得分的barplot,生存时间的point图(红色代表死亡,蓝色代表存活),4个lncRNA的表达模式。(三连图)发现在低风险组患者中,ENSG00000271797(单cox中的保护因素)更趋向于高表达;其余三个lncRNA(单cox中的危险因素)在高风险组中更趋向于高表达;同时,dead更趋向于分布在高风险组中。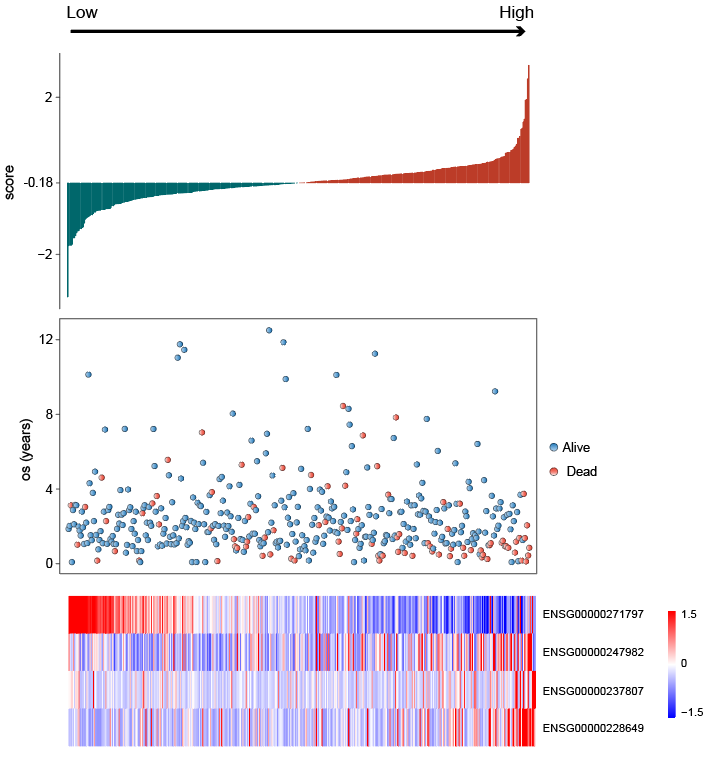
五:TCGA 的风险score和风险group分别与临床信息的独立性。
score和risk group与临床信息的单cox,使用森林图展示临床信息,HRs,95%CI 和 p-value。
score 使用的连续值,是一个危险因素,说明了score越高,风险越高。
risk group, stage 使用的是离散值,说明了high risk 对 low risk组是一个危险因素,同理,stage III/IV组对于stage I/II组也是一个危险因素。
gender 和 age对生存预后的影响并不显著。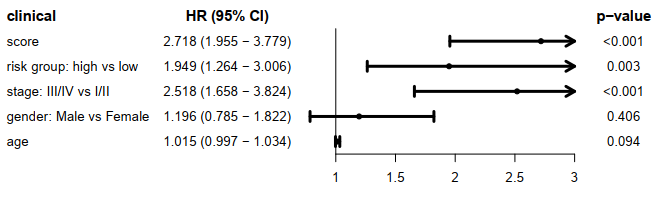
score与临床信息的多cox,使用森林图展示临床信息,HRs,95%CI 和 p-value。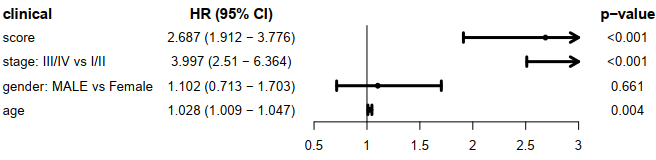
risk group与临床信息的多cox,使用森林图展示临床信息,HRs,95%CI 和 p-value。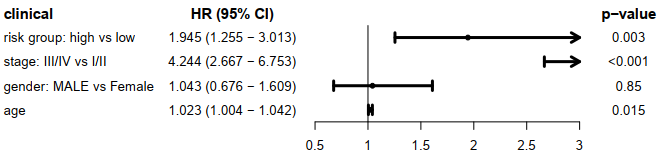
六:4个lncRNA的功能注释。在全体TCGA的COAD集合上计算4个lncRNA的表达水平和mRNA表达水平的pearson相关系数,筛选出与4个lncRNA显著相关的mRNA进行功能注释。根据三联图中lncRNA的表达模式与score的关联,选择与ENSG00000271797显著正相关,以及与ENSG00000237807, ENSG00000247982, ENSG00000228649显著负相关的mRNA进行功能注释,最终筛选得到3253**个mRNA。(p<0.05, |cor|>0.2**)
使用R的clusterprofiler包的bitr()进行ID转换,把gene symbol转换成ENTREZID。
enrichGO()进行GO_bp(Biological process)的功能注释,数据库选用的是OrgDb = “org.Hs.eg.db”,p值使用的矫正方法是FDR矫正,keyType选择的是 “ENTREZID”。
最后使用dotplot()函数,进行GO富集结果的展示,图中的count代表富集的基因书,颜色代表p值的范围。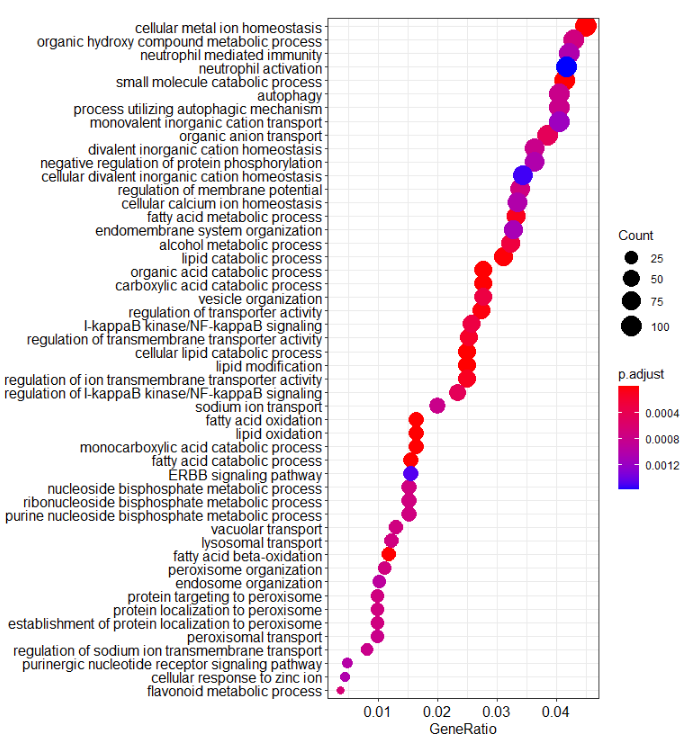
enrichKEGG()进行KEGG pathway的功能注释,pvalueCutoff选择的是0.05,p值使用的矫正方法是BH方法矫正,qvalueCutoff选择的是 0.05。
最后使用dotplot()函数,进行KEGG富集结果的展示,图中的count代表富集的基因书,颜色代表p值的范围。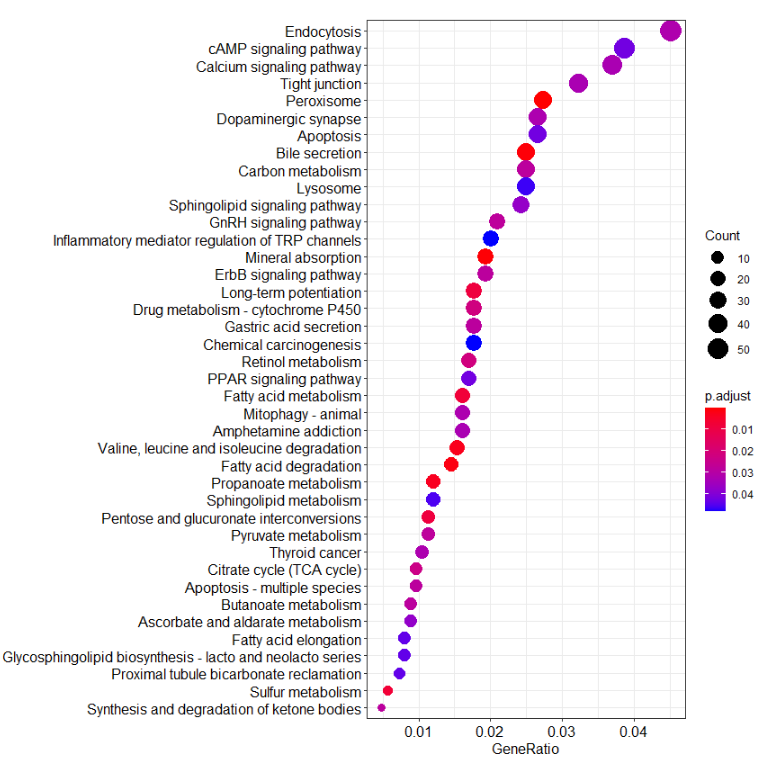
七:GEO 外部独立验证集
GSE17538。TCGA训练集中筛选的4个预后相关的lncRNA在GSE17538中全部都可以map上,因此进行风险模型的验证,风险得分的cutoff使用GEO17538集合自己的风险得分的中位值,cutoff(-0.1805057**),风险得分大于cutoff的定义为高风险组,风险得分小于cutoff的定义为低风险组。**独立验证集中KM曲线的log-rank p-value=0.08。低风险组患者的预后显著优于高风险组患者。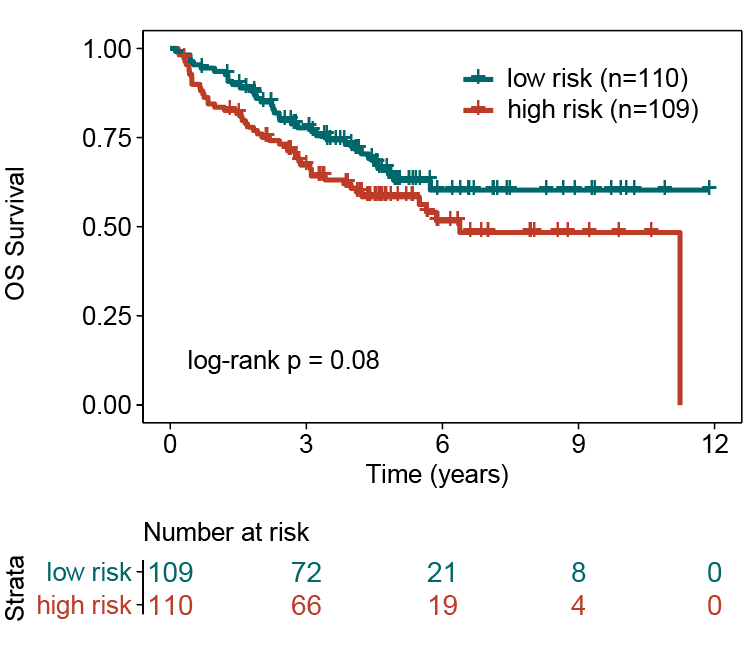
low risk:三年生存率:0.673 五年生存率:0.584
high risk: 三年生存率:0.777 五年生存率:0.630
为了体现风险模型如何预测数据集中的受试者的存活时间。特别是,假设我们有几天的生存时间,我们想看看标记如何预测三年/五年的存活(predict.time=3 years or 5 years)。该功能返回感兴趣的时间点的标记值、TP(真阳性)、FP(假阳性)、对应于感兴趣时间点(predict.time)和AUC(ROC)曲线下面积的Kaplan-Meier生存估计。使用R语言survivalROC进行上述操作方法。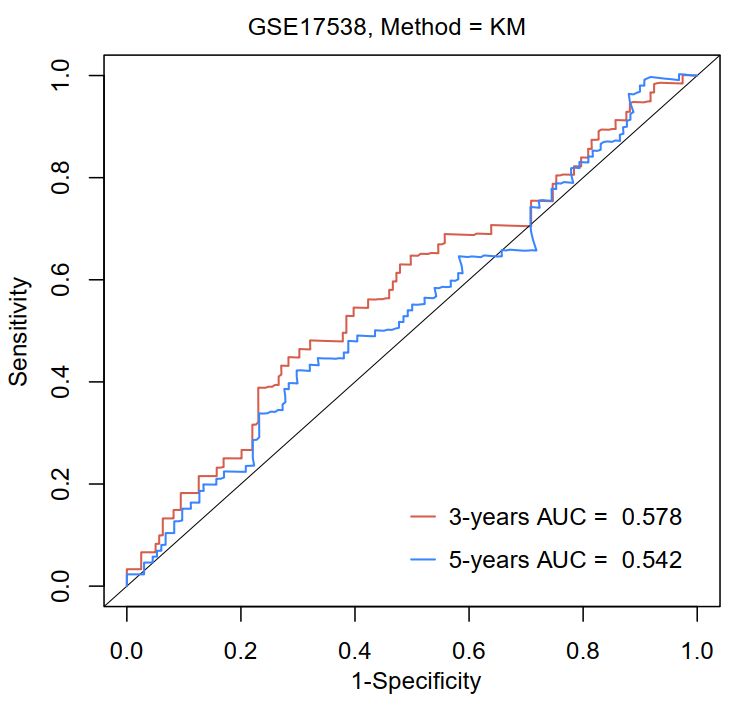
根据风险得分在训练集进行从低到高的排序,绘制得分的barplot,生存时间的point图(红色代表死亡,蓝色代表存活),4个lncRNA的表达模式。(三连图)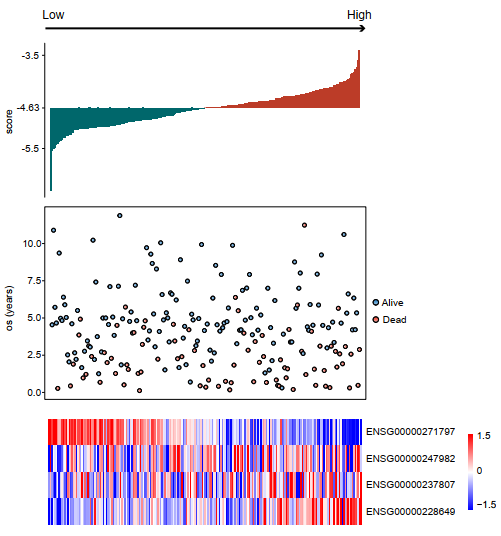
风险score和风险group分别与临床信息的独立性。
score + risk group与临床信息单cox森林图,展示临床信息,HRs,95%CI 和 p-value。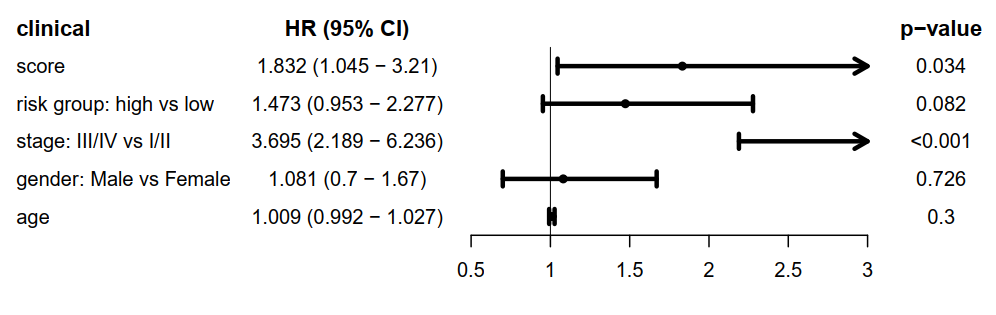
score与临床信息的多cox,使用森林图展示临床信息,HRs,95%CI 和 p-value。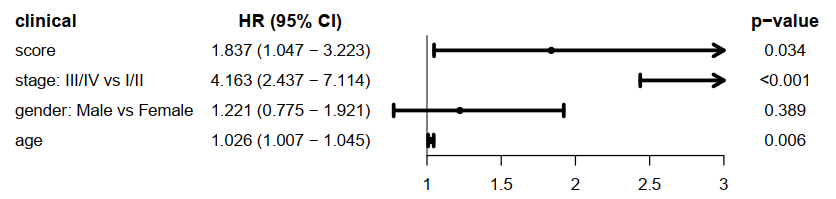
risk group与临床信息的多cox,使用森林图展示临床信息,HRs,95%CI 和 p-value。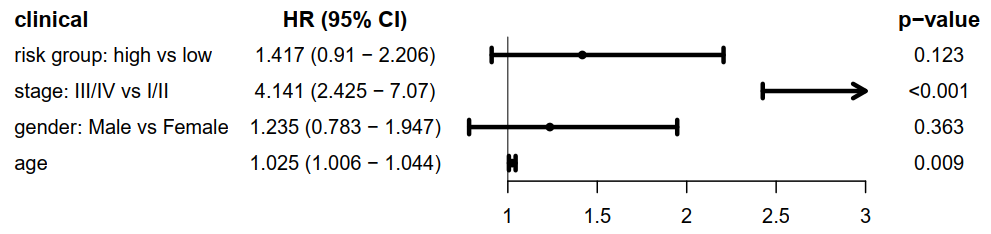

若有收获,就点个赞吧
0 人点赞
