all_mutation瀑布图 ######
setwd(“F:\ESCC\chen_data\DSR+SSR结果\未转移的病人”)
library(“optparse”)
library(“openxlsx”)
option_list = list(
make_option(c(“-f”, “—file”), type=”character”, default=”ESCC_mutation_weizhuanyi.xlsx”,
help=”输入文件名, 默认是filter_snv_indel.xlsx”, metavar=”character”),
make_option(c(“—top”), type=”numeric”, default=20,
help=”展示top多少的基因, 默认是20”, metavar=”character”),
make_option(c(“—anno”), type=”character”, default=FALSE,
help=”注释文件”, metavar=”character”),
make_option(c(“—gene”), type=”character”, default=FALSE,
help=”目标基因文件”, metavar=”character”),
make_option(c(“—keepGeneOrder”), type=”character”, default=FALSE,
help=”是否按照目标基因排序”, metavar=”character”),
make_option(c(“—showBarcodes”), type=”character”, default=”FALSE”,
help=”是否展示样本名称, 默认不展示”, metavar=”character”),
make_option(c(“—width”), type=”numeric”, default=8,
help=”是否展示样本名称, 默认不展示”, metavar=”numeric”),
make_option(c(“—outname”), type=”character”, default=”oncoplots”,
help=”输出名程, 默认是oncoplots”, metavar=”character”)
);
opt_parser = OptionParser(option_list=option_list)
opt = parse_args(opt_parser)
annovarToMaf = function(data){
lvls = c(‘synonymous SNV’, ‘nonsynonymous SNV’, ‘stopgain’, ‘stoploss’, ‘frameshift insertion’, ‘frameshift deletion’, ‘nonframeshift insertion’,
‘nonframeshift deletion’, ‘Intron’, ‘IGR’, ‘Splice_Site’, “3’UTR”, “3’Flank”, “5’UTR”, “5’Flank”, “unknown”, “UNKNOWN”, ‘RNA’, ‘Amp’, ‘Del’, ‘TMB’, “Fusion”, “SV”)
lbls = c(‘Silent’, ‘Missense_Mutation’, ‘Nonsense_Mutation’, ‘Nonstop_Mutation’, ‘Frame_Shift_Ins’, ‘Frame_Shift_Del’, ‘In_Frame_Ins’,
‘In_Frame_Del’, ‘Intron’, ‘IGR’, ‘Splice_Site’, “3’UTR”, “3’Flank”, “5’UTR”, “5’Flank”, “UNKNOWN”, “UNKNOWN”, ‘RNA’, ‘Amp’, ‘Del’, ‘TMB’, “Fusion”, “SV”)
names(lbls) = lvls
data = trim(data)
data$ExonicFunc.refGene = as.character(lbls[as.character(data$ExonicFunc.refGene)])
data.del = data[data$Alt %in% “-“, ]
data = data[!data$Alt %in% “-“, ]
if(nrow(data.del) > 0){
data.del$var.type = ‘DEL’
}
data.ins = data[data$Ref %in% “-“, ]
data = data[!data$Ref %in% “-“, ]
if(nrow(data.ins) > 0){
data.ins$var.type = ‘INS’
}
if(nrow(data) > 0){
data$var.type = ‘SNP’
}
data = rbind(data, data.del, data.ins, fill = TRUE)
Tumor_Seq_Allele2 <- data[c(“Alt”)]
data<- cbind(data, Tumor_Seq_Allele2)
colnames(data) <- c(“Tumor_Sample_Barcode”, “Chromosome”, “Start_Position”, “End_Position”,
“Reference_Allele”, “Tumor_Seq_Allele1”, “i_TumorVAF_WU”, “Hugo_Symbol”, “i_transcript_name”, “Protein_Change”, “Variant_Classification”,
“Variant_Type”, “Tumor_Seq_Allele2”)
data=data%>%filter(is.na(Tumor_Sample_Barcode)==FALSE)
data = data[!data$Tumor_Sample_Barcode %in% “TRUE”, ]
data = data[!data$Variant_Type %in% “TRUE”, ]
return(data)
}
require(maftools)
require(‘NMF’)
require(‘corrplot’)
library(dplyr)
library(raster)
data <- read.xlsx(opt$f)
data = annovarToMaf(data)
if(opt$gene != FALSE){
gene = read.xlsx(opt$gene)
gene = intersect(gene$Gene, unique(data$Hugo_Symbol))
}else{
gene = NULL
}
if(opt$keepGeneOrder != FALSE){
keepGeneOrder = TRUE
}else{
keepGeneOrder = FALSE
}
if(opt$top >= 30){
height = opt$top / 5
}else{
height = 6
}
sample_number = length(unique(data$Tumor_Sample_Barcode))
if(sample_number >= 30){
width = sample_number / 3
}else{
width = 8
}
if(opt$showBarcodes==”FALSE”){
showBarcodes=FALSE
}else{
showBarcodes=TRUE
height = height + 1
}
if(opt$anno != FALSE){
anno <- read.xlsx(opt$anno)
laml = read.maf(maf = data,removeDuplicatedVariants = F, clinicalData=anno)
pdf(paste(opt$outname, “.pdf”,sep=’’), height=height, width=opt$width)
oncoplot(maf = laml, clinicalFeatures=colnames(anno)[2:length(colnames(anno))],annotationDat=anno, sortByAnnotation=TRUE, sampleOrder=anno[[1]], writeMatrix = TRUE, removeNonMutated = FALSE, showTumorSampleBarcodes=showBarcodes, top=opt$top, genes=gene, keepGeneOrder=keepGeneOrder)
dev.off()
}else{
print(data)
laml = read.maf(maf = data,removeDuplicatedVariants = F)
print(laml)
print(gene)
pdf(paste(opt$outname, “.pdf”,sep=’’), height=height, width =opt$width)
oncoplot(maf = laml, SampleNamefontSize=0.7, showTumorSampleBarcodes=TRUE, removeNonMutated = FALSE, top=opt$top, genes=gene, writeMatrix = TRUE, keepGeneOrder=keepGeneOrder)
dev.off()
}
xlsx的文件格式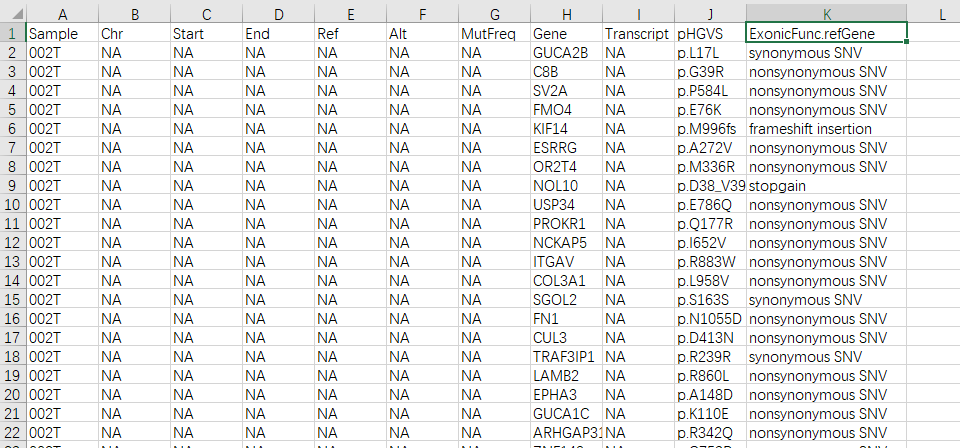
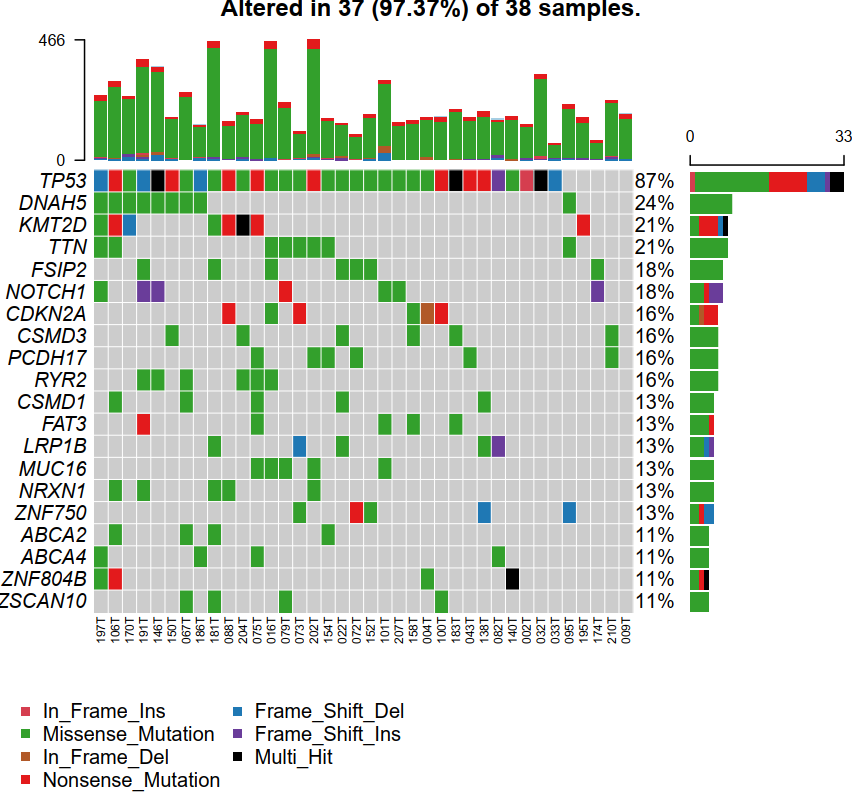
in_frame 是编码框没有移动的意思, 就是插入或者缺失碱基个数都是3的倍数
frame_shift 是编码框发生移动
ins 插入; del 缺失
missense 是单碱基错义突变
nonsense 是无义突变,就是突变产生新的终止子

若有收获,就点个赞吧
0 人点赞
